Houdini и 3D модель вируса SARS-CoV-2
Мы создали атомарную 3D модель вируса SARS-CoV-2. И хотим рассказать о нашем проекте.
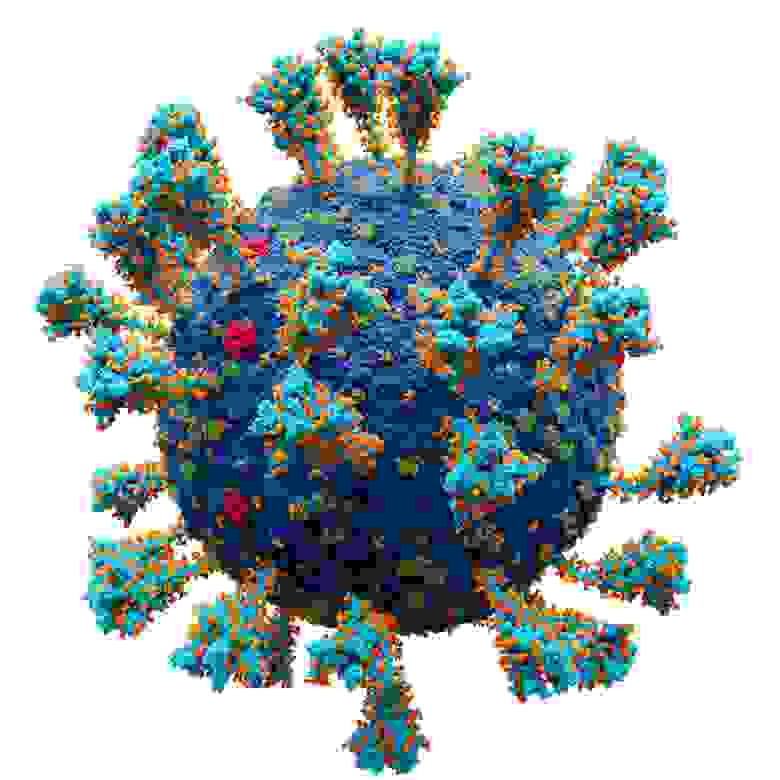 Атомарная 3D модель (каждая сфера — атом) вируса SARS-CoV-2
Атомарная 3D модель (каждая сфера — атом) вируса SARS-CoV-2
Это переработанная версия нашей статьи для N+1.
https://nplus1.ru/blog/2021/07/29/sars-cov-2-model
На иллюстрации конечный результат. Но что ему предшествовало? На просторах Сети изображений вируса на сегодняшний день весьма много. И зачем нужно ещё одно?
Пример иллюстраций по запросу в google SARS-CoV-2. В выборку попали сечения и настоящие фотографии вируса. Поэтому розовыми точками отмечены результаты поиска с рукотворными изображениями.
 Результат поиска по ключевому слову SARS-CoV-2
Результат поиска по ключевому слову SARS-CoV-2
Визуально и колористически это очень разные иллюстрации, хотя они и изображают один и тот же объект.
Для нас важна визуальная часть того, что мы делаем. Заинтересовать. Увлечь. Вызвать любопытство. Вызвать эмоции.
И если для учёного, который уже выбрал свой профессиональный путь, красивая иллюстрация — приятное подтверждение собственной правоты, то для школьника, как мы надеемся, эстетика может стать критерием выбора пути.
Также для нас очень важна научная достоверность. В своих работах мы стараемся максимально опираться на научные статьи и данные. Что не всегда возможно — много неизвестного, неизученного или противоречивого. Знания (модель реальности) постоянно обновляются и публикуются новые статьи, зачастую опровергающие ранние результаты или предположения.
Мы подумали, что можем совместить оба эти подхода. Эстетику и науку: соблюсти достоверность и одновременно сделать изображение интересным. Показать и сложность, и красоту.
Так у нас появилось желание сделать свою модель вируса.
Строение вируса и источники данных
Мы не планировали делать капсид (внутреннюю часть вируса), так как она ещё мало изучена и в научных публикациях по большей части выдвинуты предположения, чем конкретные результаты. Например, точная укладка всей полипептидной цепи N-белка, с которым упакована РНК внутри капсида, ещё не расшифрована и не смоделирована в 3D на достойном уровне. В статье можно увидеть диаграмму степени «неупорядоченности» всех фрагментов N-белка, и три из пяти фрагментов по последним данным достаточно хаотично упакованы в пространстве (выделено розовым цветом).
 Диаграмма степени «неупорядоченности» всех фрагментов N-белка
Диаграмма степени «неупорядоченности» всех фрагментов N-белка
Поэтому всю работу мы вели с внешней, видимой частью вируса.
Мы обратились к Protein Data Bank, откуда взяли часть структурных белков.
Какие это белки?
Самые хорошо изученные, оружие вируса №1, S-белки (бирюзовые), они же спайк-белки или белки-шипы.Ими вирус прикрепляется к рецепторам клетки. Если эти белки свою работу делают хорошо, то вирус пробирается в клетку и приступает к самокопированию внутри неё. В базе PDB это белок под кодовым номером 6VYB.
M-белки (зелёные) отвечают за контроль сборки новых вирионов в клетках-хозяевах. Этот белок представляет собой своеобразный каркас, на котором организуются в новый вирион другие структурные белки, а также помогает «новорождённому» вириону выбраться из клетки. На момент нашей работы этого белка не было в базе. Поэтому мы взяли похожий белок.
Е-белки (красные), взаимодействуют с другими структурными белками, а также, будучи трансмембранными белками, образуют ионный канал, тем самым помогая сборке новых вирионов в комплексе Гольджи клетки-хозяина. В базе PDB это белок имеет код 5×29. Вот так выглядит структура E-белка.
Олигосахаридные остатки (оранжевые) выступают«камуфляжем» для вируса. Вирус, прикрываясь ими, прячется от иммунной системы организма — клеткам иммунитета сложнее узнать обвешанного сахарами пришельца и, соответственно, начать выработку антител против него. В формате pdb файла S-белок содержит множество олигосахаридных белков. Удалив сам шип, мы получили экземпляр олигосахарида, который затем гликозилировали внешнюю оболочку вируса.
Точного липидного состава оболочки коронавируса в полном виде в библиотеке PBD нет. Однако несложно предположить, что состав должен быть близок к липидным оболочкам наших, человеческих, клеток. Мы использовали липиды (часть структуры 2MLS), и построили оболочку коронавируса самостоятельно. Вирус собирает свою оболочку из фрагментов мембраны клетки-хозяина — этот суперкапсид способствует повышению заразности вируса. Суперкапсид нашего вируса построен из липидов мембраны клетки эпителия.
С начала 2020 года человечество сфокусировалось на исследование SARS-CoV-2. В PDB есть специальный раздел, аккумулирующий все научные данные по вирусу SARS-CoV-2. Вот, например, картинка из этого раздела, иллюстрирующая соответствие зашифрованной белковой последовательности в геноме и 3д структур структурных и неструктурных белков коронавируса. Структурные белки — из них состоит вирус, находящийся в свободном полёте вне клетки. Неструктурные белки — те, которые синтезируются в клетке и слаженно обеспечиваются сборку новых вирионов, но в состав вириона не входят.
SARS-CoV-2 геном и протеины
https://cdn.rcsb.org/news/2021/figure-2.png
Формат файла pdb хранит информацию о каждом атоме. Углерод, водород, кислород, азот, сера. Поэтому, при желании, каждый атом можно сделать своего диаметра. Мы усреднили диаметр всех атомов. Визуальное представление отдельных белков мы видим ниже.
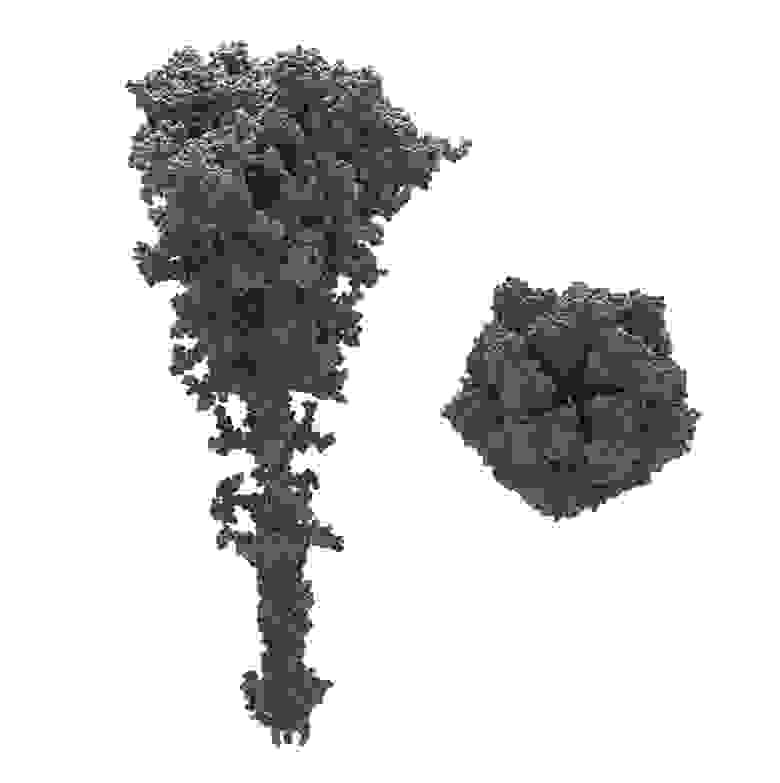 S-белок (слева) и E-белок (справа). Изображения не в реальном масштабе по отношению друг к другу
S-белок (слева) и E-белок (справа). Изображения не в реальном масштабе по отношению друг к другу
Следующим нашим шагом было определить количество объектов, из которых состоит вирус, и хаотичность или, наоборот, закономерность их расположения. Понимание того, как двигаются части и целый вирус также повышает точность модели. Вот, как выглядела наша промежуточная модель.
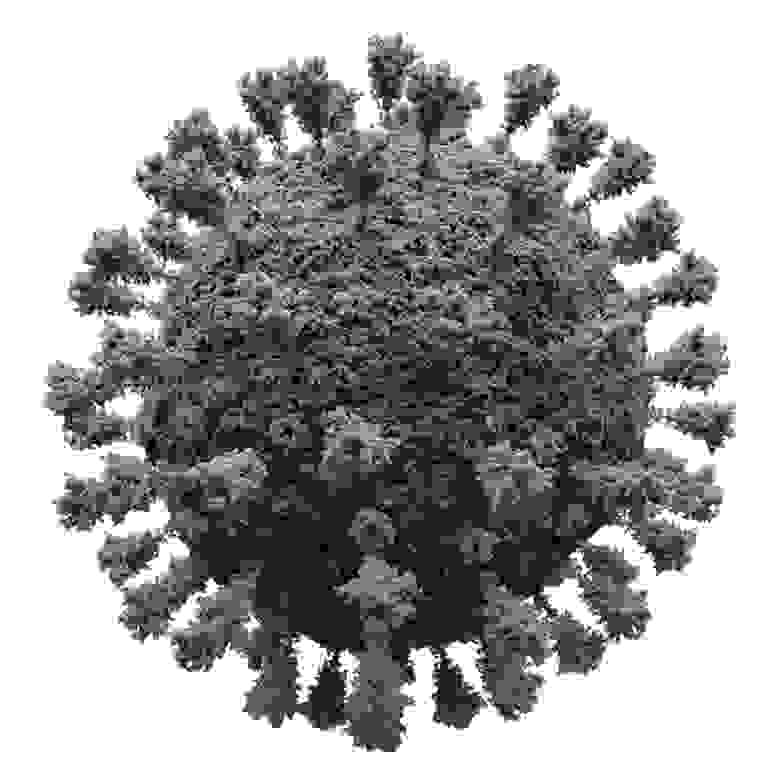 Промежуточная модель коронавируса SARS-COV-2
Промежуточная модель коронавируса SARS-COV-2
Затем нам удалось получить консультацию экспертов. Это позволило повысить точность нашей модели. Мы чрезвычайно признательны Николаю Никитину с кафедры вирусологии МГУ и Софье Борисевич из лаборатории химической физики Уфимского Института химии РАН. Вместе мы уточнили количество S-белков, зависящее от размера вириона (его размер 50–200 нанометров). По последним данным их не 90, как было в первой версии нашей иллюстрации, а меньше — от 26 до 41-го. На нашей модели S-белков 38.
Также сюрпризом для нас оказалась подвижность S-белка:
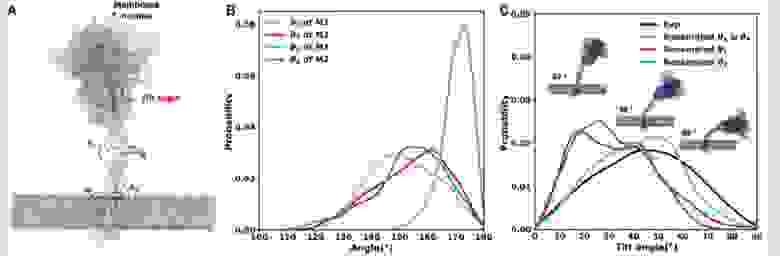 Иллюстрация, показывающая сгибы и вращение S-белка.Choi et al. / Journal of Chemical Theory and Computation, 2021
Иллюстрация, показывающая сгибы и вращение S-белка.Choi et al. / Journal of Chemical Theory and Computation, 2021
У «ножки» S-белка есть два колена, что даёт ей большую степень свободы на изгиб. Помимо наклонов, шип может ещё и вращаться вокруг продольной оси. Такая степень свободы косвенно подтверждает нашу гипотезу, что вирион не может быть плотно усеян S-белками, как было в нашей ранней модели — тогда они мешали бы друг другу. А если подвижность S-белка нарушена, то ему сложнее связаться с рецептором ACE2. Ограничение подвижности S-белков, кстати, один из способов предотвратить заражение клеток вирусами.
Помимо этого, мы выяснили, что M-белки расположены на оболочке намного плотнее — их стало около 1000 на вирион. А E-белков, как минорной компоненты, напротив, очень мало. В нашем случае их 15 штук на вирион.
Оболочка вируса, на первый взгляд, кажется очень жёстким панцирем, чем-то близким по своим свойствам корпусу подводной мины. Но это не так: её форма не является правильной сферой, а кроме того она ещё и постоянно меняется (это будет видно на анимации, она ниже). Но хотя свойства оболочки и можно представить как «жидкий кристалл», S-белки жёстко в ней закреплены, их «ножки» неподвижны.
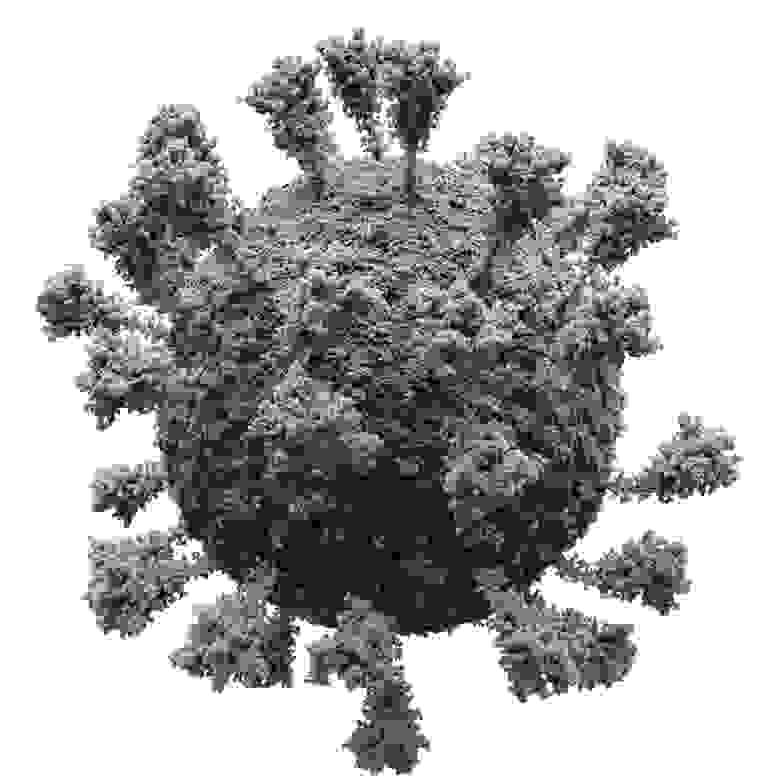 Атомарная 3D модель вируса SARS-CoV-2
Атомарная 3D модель вируса SARS-CoV-2
Мы не редуцировали количество атомов в нашей модели. Поэтому каждый белок содержит количество атомов из pdb файла. Если увеличить изображение, то можно увидеть отдельные атомы.
 Увеличенный фрагмент вируса. Хорошо видны отдельные атомы
Увеличенный фрагмент вируса. Хорошо видны отдельные атомы
Размер вириона в диапазоне от 50 до 200 нм. Это меньше длины световой волны. Поэтому в реальной жизни вирус чёрно-белый. Раскрашивают его потом.
Houdini
3D программа, в которой мы делали нашу модель. Вот как выглядит S-шип в Houdini.
 Houdini. S-белок. Каждая точка — атом
Houdini. S-белок. Каждая точка — атом
Цветом на иллюстрации показан S-белок и олигосахаридные остатки, которые плотно его облепили. Это облако точек из pdb файла. На рендере каждую точку/атом/частицу можно отобразить в виде сферы.
А вот как выглядит вирус в сборе. Houdini любезно сообщает, что в модели порядка 27 млн. атомов.
 Houdini. 3D модель вируса SARS-CoV-2
Houdini. 3D модель вируса SARS-CoV-2
По умолчанию в Houdini используется CPU рендер Mantra. Но есть возможность подключить другие внешние рендеры. Я использовал GPU рендер RedShift. Разница между AMD 3950X (16 ядер) и NVIDIA 3080 RTX в моём случае была в семь раз в пользу видеокарты. Что учитывая разнообразные тесты, а также работу с анимацией — очень существенная разница.
Вирус рендерился в чёрно-белом виде. И дальше уже по маскам красился вручную в программе для композитинга и монтажа — DaVinci Resolve. Такой подход позволил подобрать цветовую гамму без пересчёта модели. Всего было испробовано порядка десяти вариантов цветовых решений.
SARS-CoV-2 не похож на монолитное изваяние. Мы сделали анимацию, чтобы показать, как вирус «дышит» — и таким образом подчеркнуть, что это достаточно мягкая структура. Также мы старались показать движение S-белков в диапазоне углов, описанных в последних научных публикациях.
После завершения проекта
Мы понимаем, что несмотря на высокую атомарную детализацию и большую научную достоверность нашей модели нам есть куда расти. Модель можно сделать ещё точнее, ещё достовернее. О чём речь?
Вирус подвижен. При взаимодействии с клеткой, например, через ACE2 рецептор S-белок начинает трансформироваться. PDB база не хранит траектории движения белков (хотя формат файлов это позволяет). Поэтому все белки мы берём в статичном виде. И в нашем случае S-белок взят до взаимодействия с АСЕ2 рецептором.
В открытых источниках мы не нашли информации о структуре М-белка. Поэтому он показан условно. В базе данных белков SARS-CoV-2 М-белок появился, когда работа над проектом уже была завершена.
У настоящего вируса все белки, расположенные на мембране, конечно же, взаимодействуют с капсидом (внутренняя часть вируса). И эти взаимодействия наша модель не учитывает.
Суперкапсид вируса состоит из нескольких типов липидов. Мы использовали один. Его структура тесно уложена со всеми белками, которые расположены на внешней и внутренней поверхности вируса.
Более точное построение суперкапсида возможно с привлечением средств молекулярной динамики. Это требует других знаний, софта и очень мощных вычислительных ресурсов. Мы говорим о кластерах и неделях рендера. В этом случае мы получаем непросто иллюстрацию, а устойчивую (т. е. которая живёт какое-то время, а не разваливается при симуляции) анимированную модель вируса. Ниже примеры, где модель вируса получена учёными из разных лабораторий с использованием молекулярной динамики и мощных вычислительных ресурсов.
Но даже имея вычислительные мощности и специалиста по молекулярной динамике построить модель вируса на сегодня невозможно. Несмотря на то, что многие лаборатории мира сосредоточены на изучении вируса. Знания о вирусе фрагментарны, иногда противоречивы и постоянно обновляются. Это показывает наша первоначальная модель, которая, мы ещё раз подчеркнем, была основана на научных статьях, но к декабрю 2021 года уже устарела.
Вместе с тем анимация (27 млн частиц в кадре), которую мы сделали рендерится на одной видеокарте NVIDIA 3080 RTX со скоростью 1 кадр (1920×1080) за 3 минуты. Такой результат позволяет очень быстро получить визуальное представление модели. Что очень удобно для проверки гипотез и визуального отсечения явно нежизнеспособных версий вируса. При использовании честной молекулярной динамики на полноразмерном вирусе, даже без капсида, такие тесты занял бы недели на суперкомпьютерах.
Мы планируем уточнить нашу модель вируса. Заменить M-белок, а главное — добавить внутреннюю часть вируса. Показать полноценный геном из 30 000 нуклеотидов РНК и N-белков. Эта работа сложнее, что связано с фрагментарностью знаний о внутреннем устройстве вируса. Для завершения этой части проекта нам нужны знания, железо для молекулярной динамики и рендера, а главное — исследователи на краю науки, которым это так же интересно, как и нам.
Википедия
Мы решили сделать нашу иллюстрацию публичной. Доступной каждому. И передали нашу иллюстрацию Википедии.
Википедия RU
Википедия ENG
Википедия оригинал
И теперь каждый может использовать нашу работу в своих целях. Нужно лишь указать, что иллюстрация распространяется по лицензии Creative Common (CC) и авторов т. е. нас.
Список литературы
1. Surya, W., Li, Y., Torres, J. Structural model of the SARS coronavirus E channel in LMPG micelles. // Biochim. Biophys. Acta — Biomembr. — 2018. — Vol. 1860. — N. 6. — P. 1309–1317.
2. Koppisetti, R. K., Fulcher, Y. G., Jurkevich, A., Prior, S. H., Xu, J., Lenoir, M., Overduin, M., Van Doren, S.R. Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase-12. // Nat. Commun. — 2014. — Vol. 5. — P. 1–14.
3. Hillen, H. S., Kokic, G., Farnung, L., Dienemann, C., Tegunov, D., Cramer, P. Structure of replicating SARS-CoV-2 polymerase. // Nature. — 2020. — Vol. 584. — N. 7819. — P. 154–156.
4. Harris, L. J., Larson, S. B., Hasel, K. W., McPherson, A. Refined structure of an intact IgG2a monoclonal antibody. // Biochemistry. — 1997. — Vol. 36. — N. 7. — P. 1581–1597.
5. Noreng, S., Bharadwaj, A., Posert, R., Yoshioka, C., Baconguis, I. Structure of the human epithelial sodium channel by cryo-electron microscopy. // Elife. — 2018. — Vol. 7. — P. 1–23.
6. Almond, A., DeAngelis, P. L., Blundell, C.D. Hyaluronan: The Local Solution Conformation Determined by NMR and Computer Modeling is Close to a Contracted Left-handed 4-Fold Helix. // J. Mol. Biol. — 2006. — Vol. 358. — N. 5. — P. 1256–1269.
7. Hurdiss, D. L., Drulyte, I., Lang, Y., Shamorkina, T. M., Pronker, M. F., van Kuppeveld, F.J. M., Snijder, J., de Groot, R.J. Cryo-EM structure of coronavirus-HKU1 haemagglutinin esterase reveals architectural changes arising from prolonged circulation in humans. // Nat. Commun. — 2020. — Vol. 11. — N. 1. — P. 1–10.
8. Yan, Renhong, Yuanyuan Zhang, Yaning Li, Lu Xia, Yingying Guo, Q.Z. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. // Science (80-.). — 2020. — Vol. 3. — N. 3. — P. 1–8.
9. Javitt, G., Khmelnitsky, L., Albert, L., Bigman, L. S., Elad, N., Morgenstern, D., Ilani, T., Levy, Y., Diskin, R., Fass, D. Assembly Mechanism of Mucin and von Willebrand Factor Polymers. // Cell. — 2020. — Vol. 183. — N. 3. — P. 717–729.e16.
10. Daniel Wrapp, Nianshuang Wang, Kizzmekia S. Corbett, Jory A. Goldsmith, Ching-Lin Hsieh, Olubukola Abiona, B.S. G., McLellan, and J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. // Science (80-.). — 2020. — Vol. 21. — N. 1. — P. 1–9.
11. Wang, M. Y., Zhao, R., Gao, L. J., Gao, X. F., Wang, D. P., Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. // Front. Cell. Infect. Microbiol. — 2020. — Vol. 10. — N. November. — P. 1–17.
12. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular Architecture of the SARS-CoV-2 Virus. // Cell. — 2020. — Vol. 183. — N. 3. — P. 730–738.e13.
13. Oostra, M., de Haan, C.A. M., de Groot, R. J., Rottier, P.J. M. Glycosylation of the Severe Acute Respiratory Syndrome Coronavirus Triple-Spanning Membrane Proteins 3a and M. // J. Virol. — 2006. — Vol. 80. — N. 5. — P. 2326–2336.
14. B.W. Neuman, M.J. B. Supramolecular Architecture of the Coronavirus Particle. // Adv. Virus Res. — 2020. — Vol. 96. — P. 1–27.
15. Neuman, B. W., Kiss, G., Kunding, A. H., Bhella, D., Baksh, M. F., Connelly, S., Droese, B., Klaus, J. P., Makino, S., Sawicki, S. G., Siddell, S. G., Stamou, D. G., Wilson, I. A., Kuhn, P., Buchmeier, M.J. A structural analysis of M protein in coronavirus assembly and morphology. // J. Struct. Biol. — 2011. — Vol. 174. — N. 1. — P. 11–22.
16. Yu, A., Pak, A. J., He, P., Monje-Galvan, V., Casalino, L., Gaieb, Z., Dommer, A. C., Amaro, R. E., Voth, G.A. A multiscale coarse-grained model of the SARS-CoV-2 virion. // Biophys. J. — 2021. — Vol. 120. — N. 6. — P. 1097–1104.
17. Yao, H., Song, Y., Chen, Y., Wu, N., Xu, J., Sun, C., Zhang, J., Weng, T., Zhang, Z., Wu, Z., Cheng, L., Shi, D., Lu, X., Lei, J., Crispin, M., Shi, Y., Li, L., Li, S. Molecular architecture of the SARS-CoV-2 virus. // Cell. — 2020. — Vol. 183. — N. 3. — P. 730–738.
18. Choi, Y. K., Cao, Y., Frank, M., Woo, H., Park, S. J., Yeom, M. S., Croll, T. I., Seok, C., Im, W. Structure, Dynamics, Receptor Binding, and Antibody Binding of the Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein in a Viral Membrane. // J. Chem. Theory Comput. — 2021. — Vol. 17. — N. 4. — P. 2479–2487.
В работе использованы белки из PDB: 6vsb, 2mls, 5×29, 6yyt.
Алексей Солодовников (Houdini artist)
Валерия Архипова (Molecular biologist)
