Биоинформатика на минималках. Почему блохи родственники ледничков?
Актуальность тренировки.
Важный этап в развитии классификации отряда блох связан с развитием генномолекулярных исследований. В настоящее время благодаря этим методам подтверждается родственность отрядов блох и мекоптер. Активно изучаются гены рибосомальной ДНК, фактор элонгации белкового синтеза и митохондриального гена цитохромоксидаза II (COII), которые изучены у представителей большинства семейств отряда мекоптер, а также 128 видов 83 родов блох из 16 семейств (С.Г. Медведев. 2009; Whiting 2002; Whiting et al. 2008). На основе данных по этим генам различными методами построения филогенетических деревьев и моделями подсчёта попарных расстояний реконструируется филогения блох, представленная за частую в виде консенсусного древа (strick consensus) (Whiting et al. 2008). Однако, в ряде случаев, результаты генномолекулярных исследований противоречат схеме родственных связей, полученных на основе филогенетического анализа. К тому же в соответствии с различными трактовками молекулярно-генетических данных могут по-разному трактоваться происхождения ключевых инфраотрядов блох. Так с точки зрения разных авторов одни и те же клады блох могут являться как полифилетическими, так и монофилетическими группами (Whiting et al. 2008, С.Г. Медведев. 2019). Это может быть связано как с особенностями экологии современной фауны блох, так и с разного рода методическими проблемами работы с молекулярно-генетическими данными. В отношение первого пункта следует подчеркнуть, что обособление отдельных филетических линий блох, имеющих равный статус в таксономии, происходило в разные исторические промежутки времени. Это в свою очередь означает, что фауна блох являет собой разрозненные фрагменты обширной палеофауны, к настоящему моменту в значительной степени вымершей (С.Г. Медведев. 2009, С.Г. Медведев. 2019). Из этого следует, что при разных методических подходах к генно-молекулярным исследованиям мы можем упустить из вида конвергентную эволюцию нуклеотидных последовательностей (гомоплазия), что может привезти к ошибкам в определении топологии дерева и к низкой статистической поддержке ветвей. Так, например, используя данные, которые имеют разное отношение для разных таксонов и соответственно разное отношение к разным участкам генов, мы обнаружим пробелы в последовательностях, и отсутствие данных по разным локусам для разных образцов (С.Г. Медведев. 2019, Лукашов В.В. 2006). Поэтому при выборе метода исследования следует учитывать все эти проблемы и подбирать образцы наиболее изученных и распространённых таксонов блох, чтобы потом сравнить их с наиболее изученными образцами мекоптер. Таким образом мы сможем построить более достоверное дерево с высокой поддержкой.
Цель и задачи тренировки.
Целью данного тренировочного исследования являлось построение достоверного филогенетического дерева, отображающего родственное отношение отряда Siphonaptera к отряду Mecoptera на основемитохондриального гена цитохромоксидаза-II (COII).
Для достижения цели были поставлены следующие задачи:
1. Выбрать последовательности митохондриальных генов представителей родов отряда Siphonaptera в базе данных NCBI [5], в качестве ингруппы.
2. Показать на основе митохондриального гена цитохромоксидаза (COX 1) взятого у известных таксономических родов отряда Siphonaptera достоверные родственные связи друг с другом.
3. Выбрать гомологичные последовательности митохондриального гена цитохромоксидаза-II (COII) представителей из рода отряда Mecoptera близкого к отряду Siphonaptera с точки зрения филогенетического древа представленном в базе данных NCBI [1].
4. Выровнять выбранные последовательности в программе MEGA X.
5. Выбрать метод построения эволюционного дерева и модель вычисления попарных расстояний.
Материалы и методы.
Для исследования в базе данных NCBI [5] были отобраны генетические последовательности следующих шести видов блох в качестве ингруппы: Chaetopsylla globiceps (Taschenberg, 1880), Chaetopsylla trichosa (Kohaut, 1903), Ctenocephalides felis (Bouché, 1835), Ctenocephalides canis (Curtis, 1826), Ceratophyllus sciurorum (Schrank, 1803), Leptopsylla segnis (Schönherr, 1811). На основании филогенетического древа, представленного в базе данных NCBI[1], в качестве аутгруппы были аналогично отобраны генетические последовательности следующих шести видов мекоптер из рода Boreus: Boreus westwoodi (Hagen, 1866), Boreus hymalis (Linnaeus, 1767), Boreus coloradensis (Byers, 1955), Boreus nivoriundus (Fitch, 1847), Boreus brumalis (Fitch, 1847), Boreus californicus (Packard, 1870), (рис. 1.).
Эволюционная филогения была реконструирована с помощью метода UPGMA [2] и представлена в виде консенсусного древа, выведенного с помощью бутстрап-анализа из 500 реплик [3]. Выравнивание проводилось для всех последовательностей с помощью стандартного инструмента Muscle (рис. 2.) в программном обеспечении MEGA X [4]. Muscle использует прогрессивное выравнивание с горизонтальным уточнением. Пары профилей извлекаются из последовательного выравнивания и повторно выравниваются, сохраняя результаты только при улучшении объективной оценки. Выравнивание было проверено с использованием опции translate DNA to protein and vice versa (рис. 3), что позволяло оценить качество выравнивания благодаря возможности трансляции нуклеотидом в аминокислоты. Отсутствие звездочек (рис. 3.) в последовательности аминокислот, подтверждало отсутствие стоп-кодонов в аминокислотной последовательности, что свидетельствовало о правильности результатов. Не правильные результаты обычно показывают звёздочки (рис. 4). Затем выровненные последовательности были изъяты из программы MEGA для согласования. Согласование было проанализировано программным обеспечением Modeltest 3.06 PPC (Posada, D., Crandall, K.C., 1999) с использованием информационного критерия Акаике (AIC), чтобы найти наиболее обоснованную модель правдоподобия для анализа. AIC уменьшает количество ненужных параметров, которые мало влияют на описание данных, за счет штрафов за более сложные модели (Posada, D., Buckley, T.R., 2004.). Эволюционный анализ был проведен в MEGA X [4] на 64-разрядной операционной системе Windows 10 Home с 4-мя процессорами INTEL с тактовой частотой 1,6 ГГц и 1 ТБ ОЗУ. Эволюционные расстояния были вычислены с помощью метода Тадзима-Нэй [6] и выражены в единицах числа базовых замен на сайт (рис. 5.). Этот анализ включал 12 нуклеотидных последовательностей. Включенные кодонные позиции были 1-м+2-м+3-м+некодирующими. Все неоднозначные позиции были удалены для каждой пары последовательностей (опция попарного удаления). В конечном наборе данных было в общей сложности 742 позиции.
(Рис 1.)
 Вверху слева: отряд блохи. Вверху справа: отряд мекоптер. Внизу отображены семейства этих отрядов
Вверху слева: отряд блохи. Вверху справа: отряд мекоптер. Внизу отображены семейства этих отрядов(Рис. 2.)
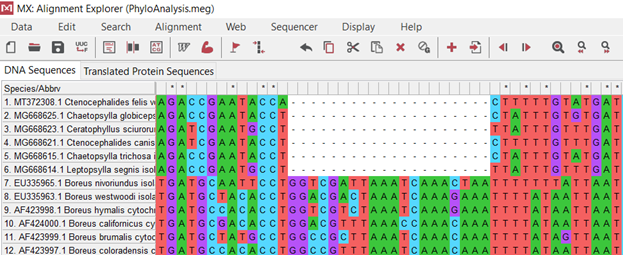 Пример произведённого мной выравнивания.
Пример произведённого мной выравнивания.(Рис. 3.)
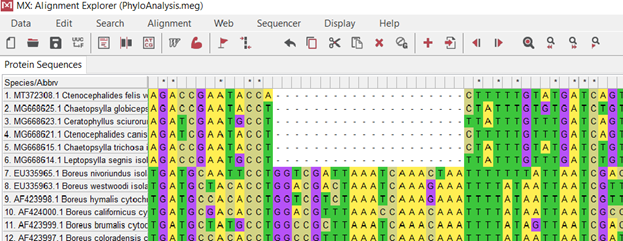 Правильный пример без звёздочек в последовательности
Правильный пример без звёздочек в последовательности(Рис. 4.)
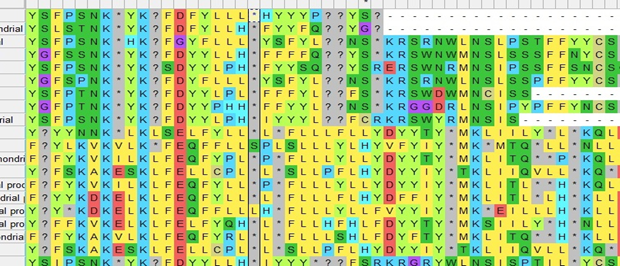 Пример со звёздочками, который мне удалось найти. Он показывает ошибку в последовательности.
Пример со звёздочками, который мне удалось найти. Он показывает ошибку в последовательности.(Рис. 5)
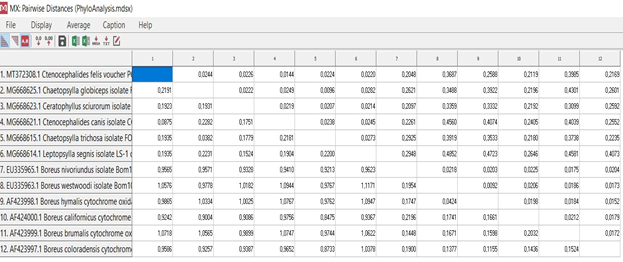 Расчёт дистанций
Расчёт дистанцийОжидаемые результаты.
Данный анализ должен показать высокую поддержку ветвей известных таксонов блох и их монофилетическое происхождение. Ожидается представление эволюционной истории всех анализируемых таксонов и поддержка гипотезы о близком родстве между отрядами Mecoptera и Siphonaptera. Поддержка общего происхождения этих двух групп выраженная в более 60% статистическом соотношении покажет состоятельность гипотезы о том, что Mecoptera является близкой сестринской группой по отношению к Siphonaptera. Напротив, низкая поддержка ветвей поставит под сомнение данную гипотезу, однако данную вероятность события возможно объяснить малой выборкой, отобранной для исследования.
Полученные результаты.
Анализ, проведённый в программном обеспечении MEGA X [4] ожидаемо показал высокую статистическую поддержку (80%-100%) для подавляющего большинство ветвей известных таксонов блох, тем самым ещё раз доказал их монофилетическое происхождение. Тем не менее для видов Ceratophyllus sciurorum и Leptopsylla segnis статистическая поддержка ветвей была выражена в меньшем соотношении (73%), однако несмотря на это она всё равно остаётся высокой и аналогичным образом подтверждает монофилетическое происхождение данных таксонов. Статистическая разница в процентах является не существенной и может объяснятся маленькой выборкой, тем не менее полученные результаты согласуются с прошлыми исследованиями по филогении блох (С.Г. Медведев. 2009; Whiting 2002; Whiting et al. 2008).
Выдвинутая гипотеза о близком родстве между отрядами Mecoptera и Siphonaptera ожидаемоподтвердилась. Поддержка общего происхождения этих двух групп выразилась для четырёх видов из двух разных таксономических групп в приближающихся ветвях. Так высокая поддержка ветвей выразилась для видов Boreus nivoriundus и Boreus brumalis (80%) и для «встречающихся» сестринских видов Chaetopsylla globiceps и Chaetopsylla trichosa (100%). Статистическое приближение двух графиков ветвей с высокой поддержкой показывает состоятельность гипотезы о том, что Mecoptera является близкой сестринской группой по отношению к Siphonaptera. Тем не менее ветка Boreus californicus имеет самую низкую поддержку ветвей (67%) на древе и здесь кажется лишней. Это может быть связано с маленькой выборкой, или с тафономическим уклоном при построении филогенетического древа. Так или иначе на результаты данная ошибка влияет не значительно и показывает состоятельность данного исследования (рис. 6.)
(Рис. 6.)
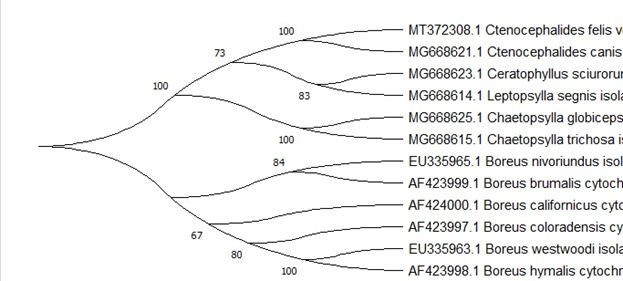 Филогенетическое дерево.
Филогенетическое дерево.Заключение.
Полученные результаты убедительно показывают эволюционную филогению групп в рассмотренном ключе. Построенное дерево принимается за представление эволюционной истории анализируемых таксонов. Дальнейшие углублённые исследования могут подтвердить, или опровергнуть озвученные выводы, поскольку генномолекулярные методы совершенствуются, а данная работа просто является тренировкой. Данное исследование никем не финансировалось и являлось пробой руки. Работа выполнена под руководством научного руководителя студенческой практики кандидата биологических наук Бабич Полины Сергеевны. Работа выполнялась самостоятельно. Работа не опубликована мной в паблике фанерозой [https://vk.com/phanerozoi] и не является копипастом. Работа не опубликована мной в данном сообществе, потому что не совсем подходит под научпоп формат.
Благодарности.
Выражаю благодарность в дельных советах и в предоставлении полезной литературы доктора биологических наук ведущего научного сотрудника ЗИН РАН Четверикова Филиппа Евгеньевича.
Выражаю благодарность в предоставлении лекционных материалов, в курировании в процессе обучения, а также к доступу к полезной литературе кандидата биологических наук Бабич Полину Сергеевну.
Выражаю благодарность в предоставлении научных работ из коллекции ЗИН РАН по филогении блох, в курировании меня в процессе освоения новых методов и советов в построение деревьев по фенотипическим признакам заведующего лабораторией паразитологии ЗИН РАН доктора биологических наук Медведева Сергея Глебовича.
Список использованной литературы1. Whiting M.F. 2002. Mecoptera is paraphyletic: multiple genes and phylogeny of Mecoptera and Siphonaptera. Zoologica Scripta, 31: 93–104
2. Whiting M.F., Whiting A.S., Hastriter M.W. and Dittmar K. 2008. A molecular phylogeny of fleas (Insecta: Siphonaptera) and host associations. Cladistics, 24: 1–31.
3. СИСТЕМАТИКА, ГЕОГРАФИЧЕСКОЕ РАСПРОСТРАНЕНИЕ И ПУТИ ЭВОЛЮЦИИ БЛОХ С.Г. Медведев, Труды Зоологического института РАН Том 313, № 3, 2009, c. 273–282
4. ОЦЕНКА РЕЛЕВАНТНОСТИ РАЗЛИЧНЫХ МОЛЕКУЛЯРНОГЕНЕТИЧЕСКИХ МАРКЕРОВ ДЛЯ ФИЛОГЕНЕТИЧЕСКОГО АНАЛИЗА ОТРЯДА БЛОХ (SIPHONAPTERA) Медведев С.Г., Илинский Ю.Ю. МАТЕРИАЛЫ III МЕЖДУНАРОДНОГО ПАРАЗИТОЛОГИЧЕСКОГО СИМПОЗИУМА СОВРЕМЕННЫЕ ПРОБЛЕМЫ ОБЩЕЙ И ЧАСТНОЙ ПАРАЗИТОЛОГИИ, 2009, с. 185–190
5. Молекулярная эволюция и филогенетический анализ/ В.В. Лукашов —М.БИНОМ. Лаборатория знаний, 2009. — с. 256. с. 29–31.
6. Posada, D., Buckley, T.R., 2004. Model selection and model averaging in phylogenetics: advantages of the AIC and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808.
7. Posada, D., Crandall, K.C., 1999. Modeltest. Bioinformatics 14, 817–818.
Ссылки
1. http://lifemap.univ-lyon1.fr
2. Sneath P.H. A. and Sokal R.R. (1973). Numerical Taxonomy. Freeman, San Francisco.
3. Felsenstein J. (1985). Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39:783–791
4. Kumar S., Stecher G., Li M., Knyaz C., and Tamura K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 35:1547–1549.
5. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4681159/
6. Tajima F. and Nei M. (1984). Estimation of evolutionary distance between nucleotide sequences. Molecular Biology and Evolution 1:269–285.
