Гражданство по ДНК, или покупают ли евреи генетические тесты
Анализ популяционной принадлежности человека по ДНК, по нашему опыту, вызывает у публики три больших вопроса: можно ли связывать между собой гены и этнические группы, как происходит анализ происхождения с технической точки зрения, и умеют ли генетические тесты «определять евреев». Почему-то именно вопрос еврейской идентичности по ДНК живо волнует как тех, кто имеет неоспоримые свидетельства о принадлежности к Б-гоизбранному народу, так и тех, кто и мацу не ест, и Тору не читает.
В новом материале Genotek на Geektimes постараемся ответить на все по порядку. И да, евреев тоже определим.

Расы aka популяционные группы в биологии, медицине и генетике
Человечество имеет вредную привычку оправдывать насилие «врожденным» превосходством одной расы над другой — оттого-то современные биологи и подходят к вопросу генетических различий между популяциями с осторожностью сапера. (Не)существование биологических границ между расовыми и этническими группами яростно обсуждалось на протяжении всего 20 века, но окончательный консенсус по этому вопросу не достигнут до сих пор (1).
Были надежды на то, что секвенирование человеческого генома всех примирит. Геном, прочитанный «от» и «до», покажет, что границы между группами имеют социальную природу, а гены одинаковы у всех. Вышло иначе: внимательное изучение нуклеотидного кода человека возродило и усилило интерес к биологическим различиям между расовыми и этническими популяциями. У одинаковых, в целом, генов нашлись слегка отличающиеся аллельные варианты, связанные с риском заболеваний (2), метаболизмом лекарств (3), ответом организма на условия окружающей среды (4), и эти варианты встречались в различных популяциях с различной частотой.
Поиск несуществующих «индийских» или «африканских» генов прекратили, однако исследования в области медицинской и популяционной генетики по-прежнему проводят параллели между биологическими особенностями и этнической принадлежностью участников. Использование терминов «раса» и «этническая принадлежность» в таких работах активно обсуждается (и нередко осуждается). Были попытки ввести правила, заставляющие исследователей обосновывать необходимость использования «скользких» категорий и уточнять, что именно понимается под конкретными терминами. В феврале прошлого года в Science, одном из самых авторитетных естественно-научных журналов, вышла неоднозначная статья (5), предлагающая и вовсе отказаться от использования термина «race» в генетических исследованиях, заменив его более корректным и нейтральным «ancestry» — «происхождение».
Но даже в условиях неопределенности с терминами делить человечество на популяционные группы по-прежнему приходится: в частности, для корректного проведения клинических испытаний лекарств и оценки риска заболеваний. Например, три аллельных варианта гена NOD2 — R702W, G908R и 1007fs — связаны с повышенным риском болезни Крона у американцев европейского происхождения (6,7), однако, ни один из этих вариантов не ассоциирован с болезнью Крона у японцев (8). Известны аллели гена CCR5, влияющие на скорость развития иммунодефицита у ВИЧ-инфицированных больных (9): среди них был найден вариант, замедляющий прогрессирование болезни у американцев европейского происхождения, но ускоряющий ее развитие у афроамериканцев (10). У азиатов обнаружили корреляцию между полиморфизмами гена белка p53, регулирующего стрессовый ответ и подавляющего развитие опухолей, и среднезимними температурами в местах обитания популяций — генетическую адаптацию к морозам (11). И если в прошлом для разбиения выборки на этнические группы использовались исключительно сведения, сообщенные самими участниками, в постгеномную эру их все чаще дополняют и уточняют генетической оценкой происхождения испытуемого.
Генетические вариации между популяциями
В повседневной жизни мы делим людей на группы по внешности или языку общения. Большинство датчан похожи друг на друга больше, чем каждый из них — на итальянца (вот классная визуализация с усредненными портретами разных народностей). Датчане и итальянцы куда ближе между собой, чем каждый из них — к жителям суб-сахарской Африки: человеческие фенотипы кластеризованы по географическому паттерну. Распределение генотипов имеет похожую структуру: члены локальной группы, как правило, имеют более тесные родственные связи, чем жители отдаленных областей, а популяции, населяющие один регион, более близки, чем те, чьи места обитания разделены географическими барьерами (например, горной грядой или водным массивом).
При этом генетическое разнообразие человеческой популяции ниже, чем у многих биологических видов. Объясняют это тем, что человечество — вид молодой: у отдельных групп было относительно немного времени, чтобы аккумулировать отличия. Два случайно выбранных человека отличаются друг от друга каждым из ~1000 нуклеотидов, тогда как два шимпанзе не совпадают раз в ~500 «букв». И, тем не менее, в сумме в человеческом геноме есть около 3 миллионов потенциальных «точек расхождения». Большинство таких несоответствий, называемых однонуклеотидными полиморфизмами (single nucleotide polymorphism, SNP), нейтрально или практически нейтрально, однако часть из них и отвечает за фенотипические отличия между людьми.
Распределение нейтральных полиморфизмов (так как они не несут биологического смысла, то и направленному эволюционному отбору не подвергаются, разносятся ветром миграций) в мировой популяции отражает демографическую историю нашего вида. Генетические и археологические данные указывают на то, что в последние 100 000 лет размер человеческой популяции значительно вырос. Люди расселялись за пределы Африки, колонизируя остальной мир. Процесс расселения влиял на географическое распределение аллелей двумя путями: во-первых, сказывался «эффект основателя» — в популяции переселенцев, как правило, была представлена только часть генетических вариантов из всего пула разнообразия их в предковой популяции; во-вторых, происходило так называемое «ассортативное скрещивание», т.е. пары образовывались преимущественно внутри своей группы, что ограничивало распространение существующих и возникающих de novo полиморфизмов среди особей, населявших различные географические области. Эти процессы вели к постепенному накоплению генетических различий.
В контексте популяционных групп геномные маркеры начали изучать в 70-х — 80-х годах, в 90-е их стали использовать для выявления популяционной принадлежности конкретного человека. Исследователи снова и снова демонстрировали, что генетические полиморфизмы позволяют успешно выделить популяционные группы и определить групповую принадлежность индивидуума. Тогда же показали, что люди, живущие на одном континенте, как правило, ближе друг к другу генетически, чем люди с различных континентов. Поначалу в таких исследованиях информация о месте рождения, расе, этнической группе была известна с самого начала и использовалась совместно с генетическими данными; если же испытуемые распределялись по кластерам «вслепую», исключительно на основе генетических признаков, соответствие между географическим происхождением, этнической принадлежностью и популяционной структурой было менее явным. Как показали дальнейшие исследования, успех зависел от используемых генетических маркеров и их количества (больше — лучше), корректного выбора референсных популяций и других факторов (12).
К 2004 году в США генетическое определение популяционной принадлежности применялось не только в биомедицинских исследованиях, но и в расследованиях преступлений: вот эта статья из Nature содержит захватывающую историю о том, как полицейские, отчаявшись найти преступника, заказали ДНК-тест в коммерческой компании, определились с цветом кожи подозреваемого и раскрыли дело. Предложения по анализу генетического происхождения удачно попали в волну повального интереса людей к собственному прошлому. «Roots mania», так назвали это увлечение в статье в Time, посвященной «America«s latest obsession» — генеалогическим исследованиям.
Активно применяют геномные методы специалисты, изучающие происхождение и эволюцию народов. Например, в 2013 году международная команда исследователей использовала генетический анализ для опровержения гипотезы происхождения евреев-ашкеназов от хазар (13). Набор геномных данных, использованный авторами, находится в открытом доступе: в нем представлено более 100 мировых популяций. Предлагаем вместе с нами смоделировать небольшое исследование: определить место клиентов Genotek в этой выборке, а заодно разобраться в технических деталях определения популяционной принадлежности.
Цель исследования
Определить место клиентов Genotek среди референсных популяций. Выяснить, есть ли в нашей выборке представители евреев-ашкеназов. Продемонстрировать принципы и методы анализа популяционной принадлежности отдельного человека.
Задачи исследования
Обработать данные генотипирования 722 испытуемых программой ADMIXTURE, используя в качестве обучающей выборки набор данных из работы Behar et al., 2013.
Материалы и методы
В исходной работе Behar et al., 2013, использовались данные 1 774 человек: среди них были представители 88-ми нееврейских популяций (из Аравии, Центральной Азии, Восточной Азии, Европы, Среднего Востока, Северной Африки, Сибири, Южной Азии и суб-сахарской Африки) и 18-ти еврейских популяций. Обширный набор данных был нужен авторам для точного определения места ашкеназов в контексте мировых популяций: стояла задача представить все три географических региона, откуда гипотетически могла произойти эта группа — Европу, Средний Восток и Хазарский каганат. Авторы особо подчеркнули разницу между подходом к отбору образцов, представляющих современные европейские, средневосточные и еврейские популяции — прямых потомков предковых популяций, и образцов, соответствующих Хазарскому каганату, который прекратил существование примерно 1000 лет назад. Загвоздка в том, что и ни одна из ныне существующих популяций не является прямой наследницей каганата. В качестве возможных современных представителей хазар авторы выбрали жителей Южного Кавказа (абхазцев, армян, азербайджанцев, грузин), Северного Кавказа (адыгов, балкарцев, чеченцев, кабардинцев, осетин и еще нескольких народностей), чувашей и татар.
Мы добавили к набору данных образцы 722 человек из различных регионов России.
Для статистического анализа использовали программу ADMIXTURE, которая позволяет оценить наиболее вероятное происхождение индивида на основании данных о генотипах. Кроме нее авторы обсуждаемой статьи применяли и другие статистические методы, давшие сходный ответ на поставленный вопрос. Мы остановимся на ADMIXTURE, так как именно этот алгоритм позволяет оценить процентный вклад предковых популяций в изучаемые геномы.
ADMIXTURE использует методы Монте-Карло в марковских цепях (Markov chain Monte Carlo, MCMC). Вот ссылка на статью авторов алгоритма для желающих подробнее разобраться в математической стороне процесса.
Рассмотрим, как работает ADMIXTURE на примере образцов и популяций из нашего набора
Всего у нас 2 496 образцов/индивидов, каждый из которых относится к одной из 106 современных популяций. Мы предполагаем, что современные популяции, скорее всего, произошли от относительно небольшого числа предковых популяций. «Предковые популяции» в таком анализе — это некие древние геномные кластеры, объединенные по принципу генетического сходства. ADMIXTURE позволяет как произвольно выдвигать предположения о числе таких кластеров в выборке, так и подбирать оптимальное их количество, наиболее корректно описывающее реальное распределение геномных данных.
Получив сведения о генотипах и предположительное количество «предковых» популяций (К), ADMIXTURE строит модель, оценивающую вклад каждой из «предковых» популяций в каждый образец. При интерпретации данных важен как количественный состав генома (процентное соотношение кластеров), так и качественный — их наличие или отсутствие в конкретных геномах. На основании этих данных можно делать предположения об эволюционных процессах в популяции, в частности, о наличии или отсутствии общих «корней» у популяционных групп. Однако выводы будут легитимны в том случае, если построенная нами модель хороша: подобрано оптимальное значение К.
Подберём оптимальное значение К
Как определить, какое количество «предковых» популяций наиболее точно соответствует истинному для данной выборки? Эмпирическим путем!
ADMIXTURE — умная программа: строя модель генетической структуры популяций на основании данных о генотипах индивидов (оценивая вклад каждого из древних геномных кластеров в каждый из геномов выборки) для заданного числа К, она не забывает в конце провести сравнение с реальностью. Проверить, насколько хорошо входные данные описываются построенной моделью. Мерой сравнения служит «ошибка» — величина, описывающая несоответствие между моделью и реальными данными. Чем больше ошибка, тем хуже предположение о количестве предковых популяций соответствует действительности.
Как выбрать оптимальное значение К? Запускаем алгоритм ADMIXTURE на данной выборке, подставляя разные значения К, и получаем для каждого К свою величину ошибки. Строим график зависимости величины ошибки от К. Вот какой график получился у авторов статьи:
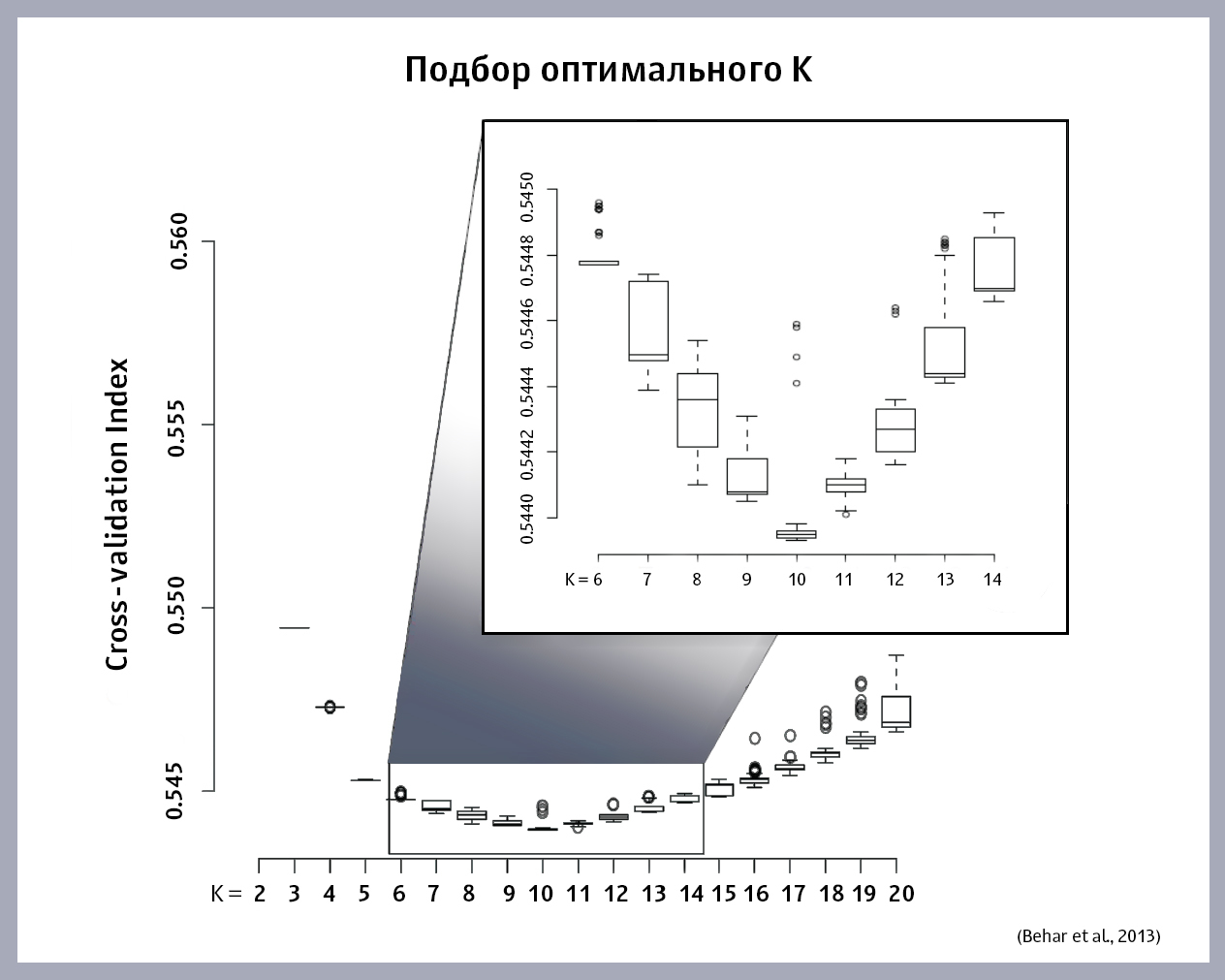
Оптимальное значение К находится в точке минимума функции. Если же минимума на графике не обнаруживается (функция постоянно растет или убывает), придется строить модели, выбирая новые К, до тех пор, пока не удастся найти нужное.
Даже при оптимально подобранном К достоверность результатов анализа зависит от корректности выборки:
1. Индивидуумы не должны быть родственниками друг другу.
2. Однонуклеотидные полиморфизмы (SNP), по которым производится генотипирование, должны быть равномерно распределены по геному с достаточно высокой плотностью.
3. Аллели SNP должны находиться в равновесном сцеплении, то есть вероятность наличия данного аллеля у конкретного индивида должно зависеть только от частоты этого аллеля в популяции, но не от других аллелей в геноме.
Как видно из графика, оптимальное К для данной выборки составило 10 «предковых» популяций.
Результаты
Результаты анализа ADMIXTURE визуализирует вот так (на рисунке видна только часть данных):
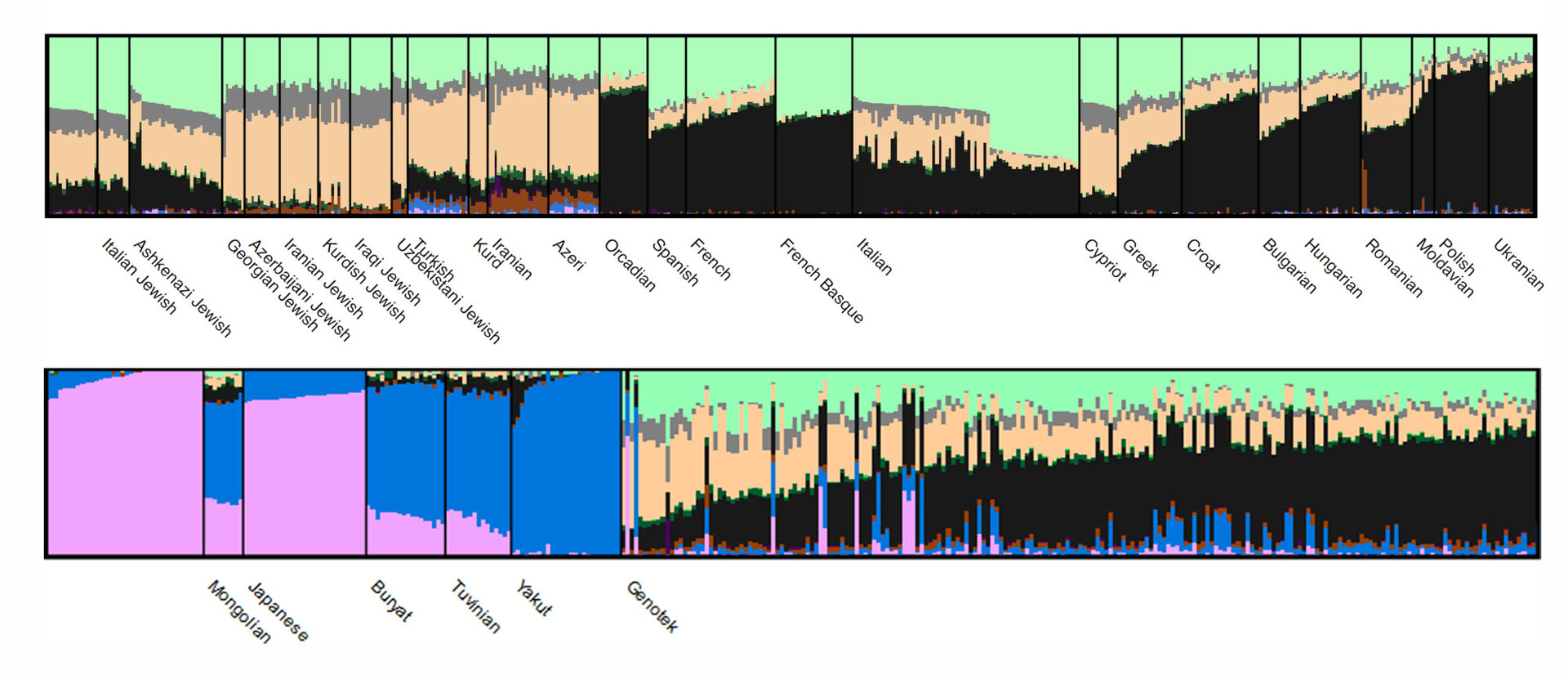
Каждому кластеру соответствует свой цвет, а популяции отличаются (или не отличаются) долями кластеров в геноме. Вот тут лежит интерактивная версия картинки для подробного изучения: наведите мышку и прокрутите, чтобы увидеть все популяции или рассмотреть какую-то из групп подробнее.
В целом, внутри «популяции» Genotek соотношение кластеров ожидаемо соответствует паттерну, характерному для популяций восточно-европейского происхождения. Интересное начинается на уровне отдельных образцов:
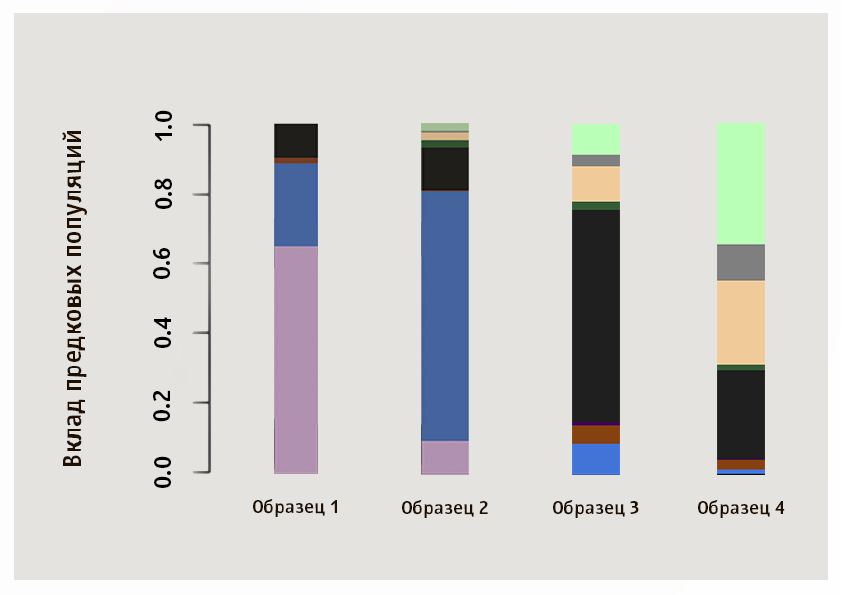
Хотя точно наиболее близкую данному образцу популяцию определяют по численным значениям, немало информации можно получить и путем визуального сравнения паттернов. Предлагаем вам самостоятельно определить наиболее близкие популяции для образцов четырех клиентов Genotek с картинки.
На этой картинке образцы 1 и 2 имеют азиатское происхождение: преобладание розового кластера характерно для японцев и и народности хан в нашей выборке, синего — для якутов, третий образец демонстрирует соотношение компонентов, характерное для русских, белорусов, украинцев и поляков, а четвертый — типичный еврей-ашкеназ.
Всего среди 722 образцов мы нашли 9 евреев-ашкеназов.
Заключение
Популяционная принадлежность — далеко не единственный фактор, определяющий этническую самоидентификацию человека. Однако выявить корреляцию между этническими группами и структурой генома их представителей все-таки возможно. Такой анализ применяется как в научных и медицинских целях, так и для изучения собственных корней всеми желающими. При этом важно понимать, что модели постоянно совершенствуются, а полученные результаты для большей точности нужно рассматривать совместно с другими данными, например, семейным генеалогическим древом.
Свидетельств хазарского происхождения ашкеназов авторами исходной статьи обнаружено не было. Генетические тесты, безусловно, «умеют» определять евреев — однако не нужно забывать, что и «еврейство» — это, прежде всего, состояние души.
В ближайшее время в Genotek запустится обновленный ДНК-тест «Генеалогия» с расширенными результатами: доведем число популяций до сотни, добавим еврейские популяции. Обновим информацию в личном кабинете для всех, кто когда-либо передавал нам свой генетический материал. Если вы все еще не отгенотипированы, приглашаем присоединиться.
Список литературы
- Foster M., Sharp R. (2002). Race, Ethnicity, and Genomics: Social Classifications as Proxies of Biological Heterogeneity. Genome Res.
- Collins F.S., McKusick V.A. (2001). Implications of the Human Genome Project for medical science. JAMA.
- Nebert D.W., Menon A.G. (2001) Pharmacogenomics, ethnicity, and susceptibility genes. Pharmacogenomics J.
- Olden K., Guthrie J. (2001). Genomics: Implications for toxicology. Mutat. Res.
- Yudell M., Roberts D., DeSalle R., Tishkoff S.(2016). Taking race out of human genetics. Science.
- Ogura, Y. et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn«s disease. Nature.
- Hugot, J.P. et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn«s disease. Nature.
- Inoue, N. (2002). Lack of common NOD2 variants in Japanese patients with Crohn«s disease. Gastroenterology.
- Martin, M.P. et al.(1998). Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science.
- Gonzalez, E. et al.(1999). Race-specific HIV-1 disease-modifying effects associated with CCR5 haplotypes. Proc. Natl Acad. Sci. USA.
- Shi, Hong et al. (2009). Winter Temperature and UV Are Tightly Linked to Genetic Changes in the p53 Tumor Suppressor Pathway in Eastern Asia. American Journal of Human Genetics.
- Bamshad M., Wooding S., Salisbury B. et al. (2004). Deconstructing the relationship between genetics and race. Nat Rev Genet.
- Behar D.M. et al. (2013). No Evidence from Genome-Wide Data of a Khazar Origin for the Ashkenazi Jews. Human Biology.
