Вирусный мегаполис: кто живет в нашем ЖКТ
Наша планета во многих аспектах уникальна, особенно если говорить про ее обитателей. На Земле обитает настолько великое многообразие видов, что даже ученые не могут прийти к общему умозаключению в оценке их числа. Кто-то считает, что видов около 5 миллионов, а кто-то смело заявляет про миллиард. Кого же на Земле больше всех? Людей, конечно, много, но даже муравьи, несмотря на свои скромные габариты, обходят нас в этом соревновании — порядка 1 квадриллиона муравьев (около 12 000 видов). Однако есть существо, которое превосходит даже муравьев. И это вирус. Группа ученых из института Сенгера (Великобритания) провели глобальное исследование, в ходе которого определили 140000 видов вирусов, обитающих в пищеварительной системе человека. О каких именно вирусах идет речь, что нового показало данное исследование, и как оно может повлиять на будущее вирусологии? Ответы на эти вопросы мы найдем в докладе ученых. Поехали.
Основа исследования
Вирусы занимают первое место по заселенности нашей планеты. По некоторым оценкам Землю населяет порядка 1031 вирионов (полноценных вирусных частиц). Вирусы способны поражать все типы организмов, от млекопитающих до бактерий. Столь широкий профессиональный профиль и является одной из основных причин внушительного видового разнообразия вирусов.
В данном исследовании идет речь о типе вирусов, которые поражают клетки бактерий и архей (одноклеточное без ядра), — бактериофаги (или сокращенно, фаги). Бактерия, которой «посчастливилось» подцепить такой вирус, испытывает весьма неприятные последствия — лизис, т.е. растворение клеток и их систем.
Вирусы это зло, сказали бы бактерии, если бы могли говорить. Но бактерии немые и не особо умные. Ибо даже негативное влияние вирусов имеет свои плюсы. Бактериофаги влияют на микробные сообщества, действуя как векторы горизонтального переноса генов, кодируя вспомогательные функции, полезные для видов бактерий-носителей. Другими словами, происходит ко-эволюционный процесс, когда вирусы развиваются из-за развития бактерий и наоборот. Как говорится, нет худа без добра.
В прошлом процесс открытия и изучения новых видов бактериофагов протекал крайне медленно. Однако с появлением новых методик, в том числе и метагеномики, все изменилось. Возможность изучать генетический материал привела к неожиданному выводу: большинство новооткрытых видов фагов нельзя было отнести к какому-либо таксону (вид, род, семейство), описанному Международным комитетом по таксономии вирусов. Это привело к тому, что каждый новый вирус определяли в той или иной группе исключительно по генетическим параметрам.
Возвращаясь к «гастрономическим» предпочтениям бактериофагов, нельзя не вспомнить про кишечник человека, в котором обитает, как мы знаем, великое множество бактерий, как полезных, так и не очень. Вполне очевидно наличие цепной реакции, когда вирус, заражающий бактерию в ЖКТ человека, влияет и на него самого, пусть даже и косвенно.
За последние годы изучение бактериофагов, обитающих в организме человека привело к множеству удивительных открытий касательно биологии фагов: липкие домены, что позволяют фагам «цепляться» за кишечную слизь; обратные транскриптазы, способствующие гипервариации генов; белки с анкириновыми доменами, которые помогают бактериям-носителям противостоять иммунитету человека.
В 2019 году было проведено исследование метагеномов (набор генов всех микроорганизмов в образце) кишечника человека, в котором было использовано около 700 образцов. За результатами этого труда была создана база данных виромов (GVD от gut virome database), т.е. совокупности вирусов, находящихся на слизистых оболочках и коже человека. Однако анализ этих данных показал, что подавляющее большинство найденных вирусов ранее не были описаны. Даже база IMG/VR, которая содержит вирусные последовательности из широкого диапазона сред, показала, что лишь 1.9% предсказаний генов* найденных вирусов были полными, а 2.5% были классифицированы как высокоточные (полные на 90%).
Предсказание генов* — определение кодирующих и регуляторных последовательностей ДНК в геноме.Следовательно, 700 образцов недостаточно для полного анализа вирусного микробиома ЖКТ человека. Именно потому и было проведено исследование, о котором мы сегодня и говорим.
Ученые собрали 28060 метагеномных образцов, полученных от людей с разных уголков планеты. В результате удалось проанализировать 142809 геномов бактериофагов, что в результате стало основой для новой базы данных кишечных бактериофагов (GPD от gut phage database).
Подготовка к исследованию
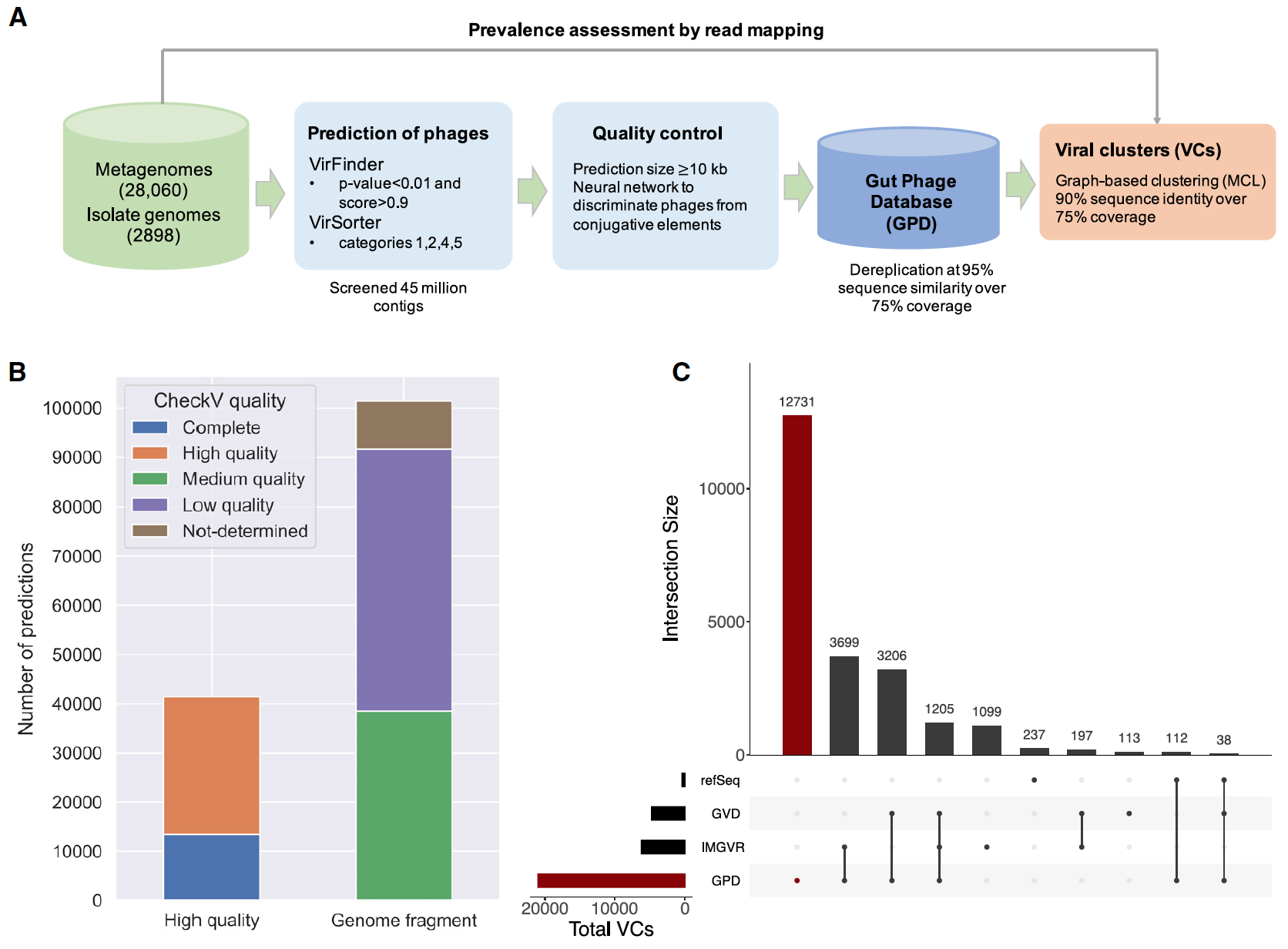
Чтобы получить полное представление о разнообразии кишечных фагов человека, было проанализировано 28060 метагеномов кишечника человека и 2898 геномов бактериальных изолятов*, культивируемых из кишечника человека (1А).
Изоляция* — в микробиологии обозначает отделение определенного штамма от естественной смешанной популяции живых микробов, присутствующих в окружающей среде. Результатом такой процедуры является изолят.
Изображение №1
Чтобы идентифицировать вирусные последовательности среди метагеномов кишечника человека, более 45 миллионов собранных контигов* были проанализированы с помощью VirFinder (исходный код), который полагается на сигнатуры k-мера* для различения вирусных контигов от бактериальных, и с помощью VirSorter (исходный код), который использует сходство последовательностей с уже известными фагами и/или другими вирусоподобными особенностями.
Контиг* — набор перекрывающихся сегментов ДНК, совокупность которых представляет собой консенсусную область ДНК.
k-мер* — последовательность из k нуклеотидов.Поскольку получение высококачественных геномов было крайне важным для последующих анализов, были использованы консервативные настройки для обоих инструментов. Потому учитывались только прогнозы длиной не менее 10 kb (kilobase — cпаренные основания*).
Спаренные основания* — пара двух азотистых оснований нуклеотидов на комплементарных цепочках нуклеиновых кислот.Для повышения точности исследования также было задействовано машинное обучение, выполняющее фильтрацию загрязненных мобильных генетических элементов* (MGE от mobile genetic element).
Мобильные генетические элементы* — последовательности ДНК, которые могут перемещаться внутри генома.Были определены предсказания генов, несущие характеристики систем секреции типа IV*. Это предполагает некую степень загрязнения конъюгативными мобильными элементами, такими как плазмиды или интегративные и конъюгативные элементы* (ICE от integrative and conjugative elements).
Система секреции типа IV* — комплекс белков секреции, обнаруженный у грамотрицательных бактерий, грамположительных бактерий и архей.
Интегративные конъюгативные элементы* (ICE) — группа хромосомно интегрированных самопередающихся мобильных генетических элементов (MGE), которые участвуют в формировании функций бактерий и бактериальных сообществ.Чтобы отличить ICE от бактериофагов, была задействована нейронная сеть с прямой связью, использующая различия в плотности генов, во фракциях гипотетических белков и в сигнатурах состава k-меров.
Оценка уровня плотности каждого вирусного генома была выполнена с помощью CheckV (1B). Этот инструмент выводит ожидаемую длину генома для предсказания вируса на основе средней идентичности аминокислот с базой данных полных вирусных геномов из NCBI (National Center for Biotechnology Information, т.е. национальный центр биотехнологической информации) и образцов окружающей среды.
В общей сложности 13429 (9.4%) вирусных геномов были классифицированы как полные, 27999 (19.6%) — высококачественные и 101381 (70.99%) — фрагменты генома (полнота менее 90%). Средняя полнота генома по всем геномам, хранящимся в GPD, была оценена в 63.5%.
Результаты анализа образцов
Чтобы оценить вирусное разнообразие GPD на высоких таксономических уровнях, использовался кластерный подход на основе графов для группировки генетически связанных фагов.
Объединение GPD с фагами из RefSeq и двумя другими базами кишечных фагов человека (GVD и IMG/VR) привело к генерации 21012 нестандартных вирусных кластеров (VC от viral cluster) с минимум одним GPD предсказанием (GPD VC).
Удивительно то, что менее 1% (171 из 21 012) VC GPD перекликаются с фагами из базы RefSeq. Фаги из этих 171 VC в основном инфицируют бактерии из родов Escherichia, Enterobacter, Staphylococcus и Klebsiella.
В соответствии с предыдущими работами по предсказанию фагов на основе наборов метагеномных данных, ученым не удалось определить большинство (80%) VC GPD к какому-то семейству вирусов. Остальная же доля относилась в основном к Podoviridae, Siphoviridae и Myoviridae. Эти семейства относятся к роду Caudovirales, для фагов которого характерно наличие хвоста и икосаэдрического капсида (двадцатигранная оболочка). Их можно в изобилии найти в фекалиях человека.
В дополнение были рассмотрены VC без GPD предсказаний (1C). Анализ VC, состоящих только из геномов, присутствующих в базах GPD и IMG/VR, показал 3699 перекрытий. А вот VC, состоящих только из геномов из баз GPD и GVD, было всего 3206. Более того, в GPD было наибольшее количество уникальных VC — 12731 кластер. Для IMG/VR и GVD также были уникальные VC — 1099 и 113, соответственно. 1205 VC присутствовали сразу в трех базах.
Уникальность созданной базы GPD заключается также и в возможности присвоить конкретный бактериофаг к его бактерии-носителю, что позволяет изучать биологию связи бактерия-фаг внутри кишечника человека.
В ходе определения бактерий-носителей было использовано 898 высококачественных геномов бактериальных изолятов кишечника человека. Благодаря данному анализу удалось отнести 40932 фага из GPD (28.66% от всех прогнозов) к 2157 штаммам-носителям.
Кроме того, анализ совместного нахождения между фагом и его предполагаемым штаммом-хозяином показал, что они обнаруживались в одном и том же метагеномном образце в 92% случаев.
Далее был проведен анализ, нацеленный на выявление какого-либо предпочтение фаговой инфекции среди 4 распространенных кишечных бактериальных филумов* человека (Firmicutes, Bacteroidetes, Proteobacteria и Actinobacteriota).
Филум* (тип) — один из высших рангов таксономической иерархии.Было обнаружено, что на уровне филума для Actinobacteriota распространенность фагов составляла 58.79%, тогда как для остальных трех бактерий — более 70%.
На следующем этапе исследования были проведены измерения вирусного разнообразия в пределах каждого из вышеперечисленных филумов. Этот анализ показал, что Firmicutes обладают значительно более высоким вирусным разнообразием (в среднем 3.13 VC на изолят), а также несут 60% от общего количества VC.

Изображение №2
Анализ на уровне рода бактерий во всех филумах показал, что Lachnospira, Roseburia, Agathobacter, Prevotella и Blautia A содержат наибольшее количество VC на изолят (2А). За исключением Prevotella, который принадлежит к семейству грамотрицательных Prevotellaceae, все эти роды являются членами грамположительного семейства Lachnospiraceae Firmicutes, ассоциированного со спорообразователями, вырабатывающими бутират. Самое же низкое вирусное разнообразие было выявлено у Helicobacter и у молочнокислых бактерий Lactobacillus H, Lactobacillus, Enterococcus D и Pediococcus.
Картина видового разнообразия вирусов показывает широкое распределение бактериофагов среди кишечных бактерий человека, даже в пределах одного и того же филума.
Ученые отмечают, что горизонтальный перенос генов между бактериями посредством трансдукции является основным двигателем потока генов в бактериальных сообществах. Следовательно, тропизм* бактериофага к носителю (бактерии) ограничен филогенетическими барьерами, при этом большинство фагов обычно ограничиваются одним видом бактерий-носителей. Другими словами, для вида каждого вируса должна быть своя бактерия.
Тропизм* — реакция ориентирования клетки, то есть направление роста или движения клеток относительно раздражителя.В рамках данного труда было установлено, что порядка 64.51% обнаруженных VC следовали тактики заражения индивидуального вида бактерий. При этом были и вирусы, которые обладали более широким диапазоном заражения: 22.39% — заражали бактерии в пределах одного рода; 10.79% — одного семейства; 1.86% — одного порядка; 0.26% — одного класса и 0.13% — одного типа (филума).
Важно то, что в рамках широкого диапазона заражения была обнаружена сильная связь между филогенетически различными видами бактерий, которая приводит к адаптационным и эволюционным процессам (2B). Другими словами, вирусы, заражающие разные виды бактерий, были на это способны по причине того, что эти разные виды между собой взаимодействовали.
Географический фактор
Как мы уже знаем, в данном труде было использовано весьма внушительное число образцов — 28060 метагеномов. География забора образцов охватывает 28 стран и 6 регионов (Африка, Азия, Европа, Северная Америка, Южная Америка и Океания). Вполне очевидно, что регион проживания может иметь немалое влияние на вирусный и бактериальный состав кишечника человека.
Действительно, анализ коэффициента Жаккара (мера сходства) показал четкое разделение североамериканских, европейских и азиатских фагеомов (бактериофаговый микробиом) от африканских и южноамериканских образцов (3A).

Изображение №3
Такая картина объясняется разным образом жизни разных людей из разных регионов. К примеру, образцы, полученные из Африки и Южной Америки, в основном отбирались из Перу, Танзании и Мадагаскара. В частности, перуанские и танзанийские образцы происходят из групп охотников-собирателей, тогда как малагасийские образцы были собраны из сельскохозяйственных групп. Исключением была Океания, так как в ней присутствовала одинаковая доля образцов, принадлежащих к обеим группам.
Ученые предположили, что бактериальный состав микробиома человека напрямую влияет на бактериофаговый состав. К примеру, бактерии Prevotellaceae более многочисленны и преобладают у людей, ведущих сельский образ жизни, тогда как Bacteroides более многочисленны и преобладают у людей, ведущих городской образ жизни.
Используя данные о назначении носителя для каждого фага, была обнаружена значительно более высокая доля VC, отнесенных к семейству Prevotellaceae из образцов метагенома из Африки, Южной Америки и Фиджи, чем из образцов из Северной Америки, Европы, Азии и Австралии (3B). Напротив, фаги Bacteroides были значительно более распространены в образцах из Северной Америки, Европы, Азии и Австралии.
Из этих наблюдений следует очевидный вывод — образ жизни человека, место его обитания (следовательно, и диета тоже) принимают непосредственное участие в формировании как бактериального, так и вирусного микробиома кишечника.
Распространение фагов в мире
Учитывая, что фаговый состав кишечника человека формируется преимущественно за счет бактериального состава, то вполне ожидаемо должна быть сильная корреляция между распространенностью VC и их бактериальным носителем.
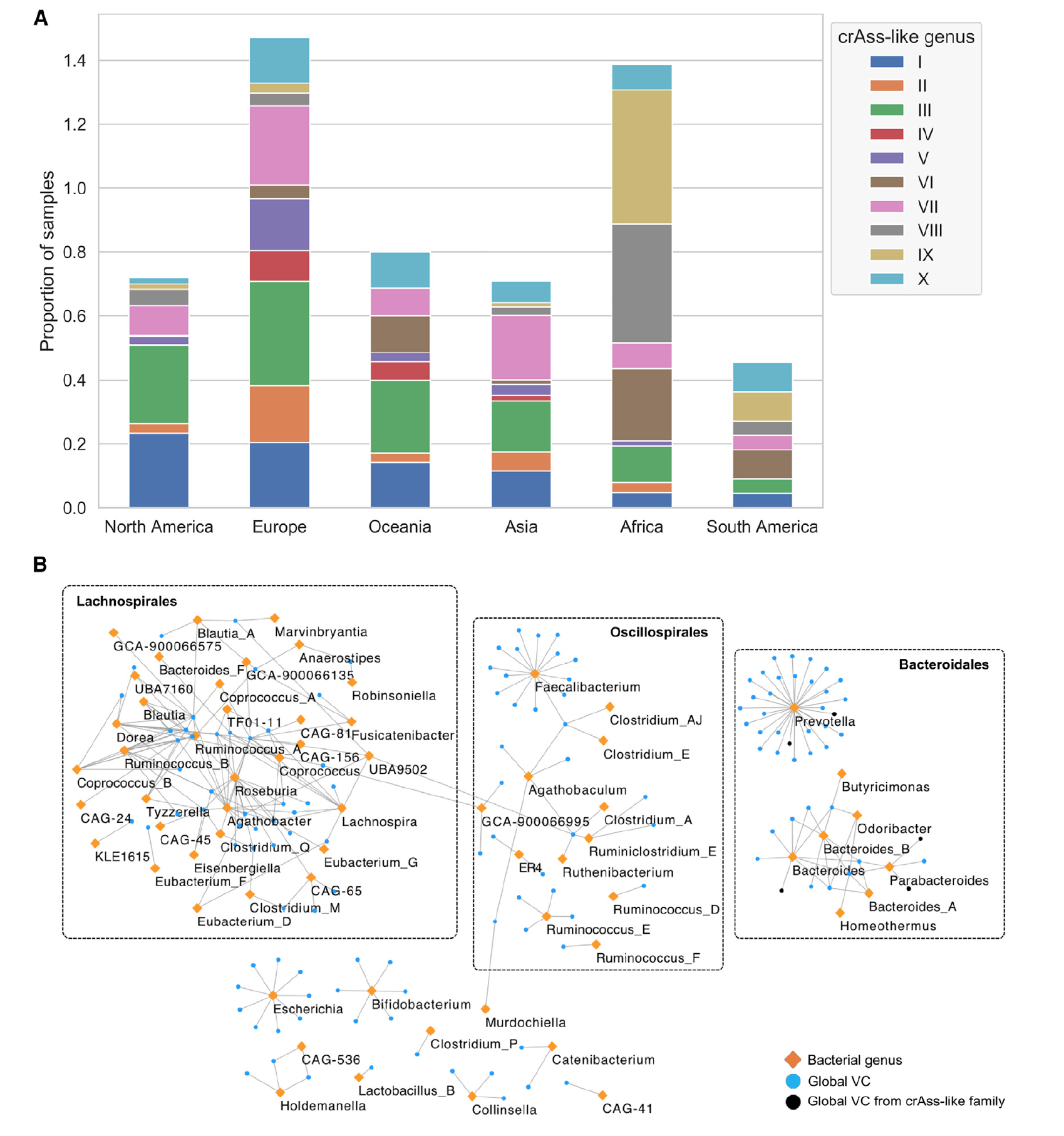
Ярким примером является crAss-подобное семейство кишечных фагов, которое можно разделить на 10 родов. Род I, который был обнаружен в значительной доли образцов из западных регионов, способен инфицировать бактериальные виды из рода Bacteroides. А вот роды VIII и IX считаются наиболее распространенными crAss-подобными фагами среди малавийских образцов. Наиболее вероятным носителем для этих двух родов бактериофагов ученые называют бактерию Prevotella copri.
Также анализ малавийских образцов показал, что распространенность родов VI, VIII и IX выше, чем у рода I в Африке, Южной Америке и на Фиджи (4A).
Изображение №4
Из этого следует, что crAss-подобное семейство глобально распределено с четко выраженными схемами распределения на уровне родов, на которые сильно влияют образ жизни человека и энтеротипы*.
Энтеротип* — классификация живых организмов на основе их бактериологической экосистемы в микробиоме кишечника.Анализ образцов выявил порядка 280 VC, распространенных по всему миру (как минимум в 5 из 6 исследуемых регионах). Эти VC составляют всего лишь 1.3% от общего числа выявленных в ходе данного исследования вирусов. Удалось определить, что 119 VC (42.5%) из 280 относятся к отряду Caudovirales.
Детальное рассмотрение идентифицированных VC показало наличие следующих семейств: Podoviridae (10 VC), Myoviridae (28 VC), Siphoviridae (43 VC) и недавно сформированное семейство Herelleviridae (1 VC).
Сеть бактерий/фагов, состоящая из глобально распределенных VC (4В), показала, что Prevotella был наиболее распространенным родом (37 VC), за ним следуют Faecalibacterium и Roseburia с 15 VC в каждом.
Также было обнаружено, что глобально распределенные фаги имеют значительно более широкий диапазон (среди разных родов) бактерий-носителей, чем фаги, обнаруженные в отдельных регионах. Следовательно, способность бактериофага заражать более широкий видовой диапазон бактерий способствует его распространению по миру.
Данный анализ показал, что наряду с двенадцатью crAss-подобными бактриофагами существует еще как минимум 280 VC, которые распространены по всему миру, вне зависимости от образа жизни или региона обитания человека.
Для более детального ознакомления с нюансами исследования рекомендую заглянуть в доклад ученых и дополнительные материалы к нему.
Эпилог
В природе не бывает самодостаточных существ, способных существовать без какого-либо влияния со стороны других видов. Это влияние может быть как постоянным, так и быть однократным толчком развития, произошедшим тысячи лет тому назад. В любом случае, утверждение, что все в мире взаимосвязано, это не гиперболизация.
Если же говорить про человека, то его тело является домом для невероятного числа самых разных микроорганизмов, начиная от бактерий и вирусов, и заканчивая грибками и археями. Этот микромир, именуемый микробиомом, играет крайне важную роль в жизнедеятельности человека. Некоторые бактерии, обитающие в ЖКТ способствуют лучшему усваиванию пищи, играют важную роль в синтезе витаминов B и K. Отношения человека и бактерий, живущих в его теле, по большей степени можно назвать симбиотическими. Однако есть и вредоносные бактерии, от которых мы всячески пытаемся избавиться, когда они портят нам здоровье.
Помимо бактерий, в нашем теле присутствуют и их злейшие враги — бактериофаги — вирусы, которые атакуют бактерии. В данном труде ученым удалось установить, что в кишечнике человека обитает порядка 140 000 видов бактериофагов.
Однако важно не только количество, но и факт того, что разные вирусы атакуют разные бактерии. Эти знания позволяют не только лучше понять влияние бактериофагов на бактерии, но и то, как эта взаимосвязь может влиять на человека. Ведь существует также и процесс ко-эволюции, когда бактерии в попытках защититься от ненавистных вирусов вырабатывают те или иные механизмы адаптации. Эти механизмы могут нести в себе как положительные, так и негативные эффекты для человека, внутри которого протекает эта борьба микроорганизмов.
Фраза «хочешь познать мир — начни с себя» отлично отображает суть данного исследования. Наше тело, словно мегаполис, является домом для колоссального числа микроорганизмов, некоторые из которых явно относятся к деструктивным элементам. Лучшее понимание микробиома позволяет нам лучше защитить себя от их пагубного влияния.
Благодарю за внимание, оставайтесь любопытствующими и отличных всем выходных, ребята! :)
Немного рекламы
Спасибо, что остаётесь с нами. Вам нравятся наши статьи? Хотите видеть больше интересных материалов? Поддержите нас, оформив заказ или порекомендовав знакомым, облачные VPS для разработчиков от $4.99, уникальный аналог entry-level серверов, который был придуман нами для Вас: Вся правда о VPS (KVM) E5–2697 v3 (6 Cores) 10GB DDR4 480GB SSD 1Gbps от $19 или как правильно делить сервер? (доступны варианты с RAID1 и RAID10, до 24 ядер и до 40GB DDR4).
Dell R730xd в 2 раза дешевле в дата-центре Maincubes Tier IV в Амстердаме? Только у нас 2 х Intel TetraDeca-Core Xeon 2x E5–2697v3 2.6GHz 14C 64GB DDR4 4×960GB SSD 1Gbps 100 ТВ от $199 в Нидерландах! Dell R420 — 2x E5–2430 2.2Ghz 6C 128GB DDR3 2×960GB SSD 1Gbps 100TB — от $99! Читайте о том Как построить инфраструктуру корп. класса c применением серверов Dell R730xd Е5–2650 v4 стоимостью 9000 евро за копейки?
