SARS нерукотворный? Генеалогия уханьского коронавируса

Не, ну какая рукотворность? Что за бред? Думал я, когда впервые услышал гипотезу о том, что Ковид-19 вызван то ли лабораторной утечкой, то ли вообще целенаправленной биоатакой. И каждый раз просто отмахивался от этих домыслов, когда они в очередной раз доплывали до меня в бурном потоке коронавирусного инфошума. Ну подумаешь, есть в Ухане институт вирусологии, мало ли.
В какой-то момент отмахиваться уже пришлось аргументированно, потому что сторонники рукотворности начали обосновывать свои тезисы о возможной искусственной природе вируса доводами из молекулярной биологии, и тут уже хотелось в пух и прах разбить их конспирологию холодными научными фактами. Уж если не как авторы статьи в Nature (казалось мне), то хотя бы как уважаемый мной Панчин.
И вот тут, в погоне за доводами против рукотворности вируса, меня и заразил вирус сомнений. В чём, собственно, причина сомнений? В том, что чем глубже погружаешься в деятельность коронавирусологов за последние 15–20 лет, тем лучше понимаешь, что создание ровно таких химер как CoV2 у них было обыденным делом. А CoV2 — это очевидная химера, основанная на летучемышином штамме RaTG13, у которого в шиповидном белке место связывания с рецептором (RBM) заменено с летучемышиного на панголиний, и вдобавок врезан особый участок из 4-х аминокислот, создавший furin cleavage site, который, как ранее выяснили вирусологи, значительно расширяет «репертуар» вируса в плане того, в чьи клетки он может проникать. Скорее всего, именно благодаря этому новому фуриновому сайту, новый мутант и сумел перескочить с исходных носителей на людей.
С учётом тех высот, которых сегодня достигла генная инженерия, синтетически собрать CoV2 по вышеописанной методике не составило бы труда даже начинающему специалисту. Ведь вирусологи, включая руководителя коронавирусного направления в Уханьском институте вирусологии Ши Чжэнли, такими вещами уже неоднократно занимались — как заменой RBM у одного вида вируса на RBM из другого (вот работа группы Ши Чжэнли от 2007 года), так и добавлением нового фуринового сайта, способного дать специфичному к одному виду животных коронавирусу возможность начать использовать рецептор ACE2 других видов.
Минутка заботы от НЛО
В мире официально объявлена пандемия COVID-19 — потенциально тяжёлой острой респираторной инфекции, вызываемой коронавирусом SARS-CoV-2 (2019-nCoV). На Хабре много информации по этой теме — всегда помните о том, что она может быть как достоверной/полезной, так и наоборот.Мы призываем вас критично относиться к любой публикуемой информации
Официальные источники
Если вы проживаете не в России, обратитесь к аналогичным сайтам вашей страны.
Мойте руки, берегите близких, по возможности оставайтесь дома и работайте удалённо.Читать публикации про: коронавирус | удалённую работу

Ши Чжэнли в своей лаборатории в Уханьском институте вирусологии
Но прежде чем разводить тут конспирологию, давайте сначала окунёмся в биологию.
Биология
Итак, начнём от печки. Что за фуриновый сайт, что за RBM, что вообще за шиповидный белок? На самом деле, если продраться сквозь дебри терминологии, то концептуально всё просто. Вот, например, шиповидный белок — это та самая торчащая из вирусной частицы штука (S protein), за которую эти вирусы и «короновали»:
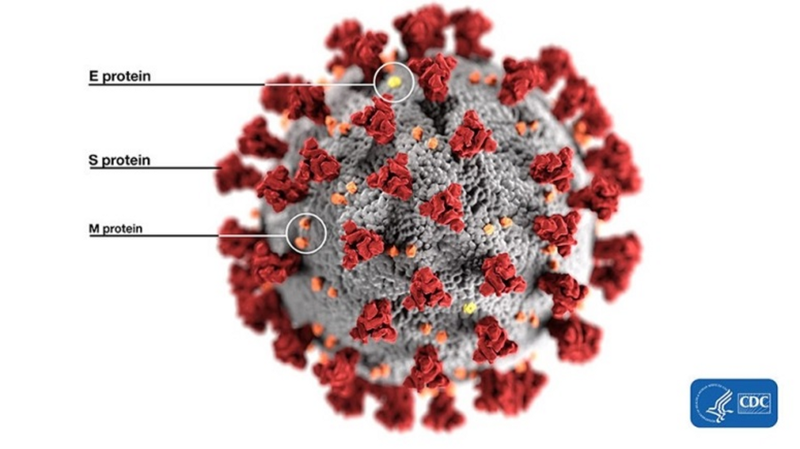
Именно с помощью этих белков вирион цепляется за рецептор клетки-жертвы (ACE2 в нашем случае), чтобы затем проникнуть внутрь. Поэтому можно сказать, что это самая важная часть вируса, ведь именно она определяет каких животных тот может поражать, а каких нет — ACE2 рецепторы у разных видов немного структурно отличаются. При этом из всего огромного по вирусным меркам 30-килобазного генома, ген этого белка составляет лишь 12–13%, то есть около 1300 аминокислот. Вот так структурирован шиповидный белок у CoV2 и его близких родственников:
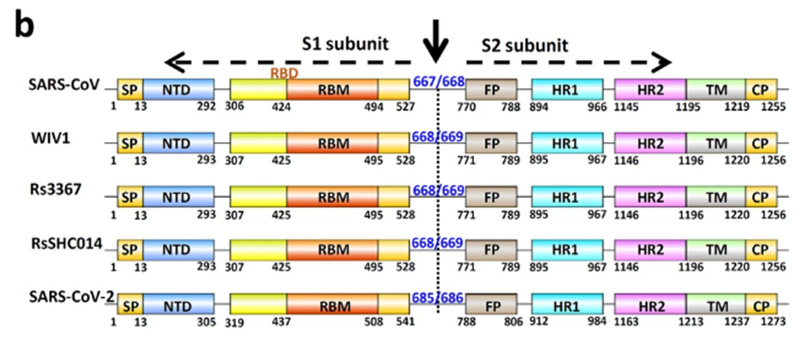
Как видно из рисунка выше, S белок состоит из двух субъединиц: S1 и S2. Именно S1 взаимодействует с рецептором ACE2, и то место, которым он это делает, называется Receptor Binding Domain (RBD), а область непосредственного контакта, святая святых, называется Receptor Binding Motif (RBM). Вот красивая иллюстрация из не менее красивой работы:
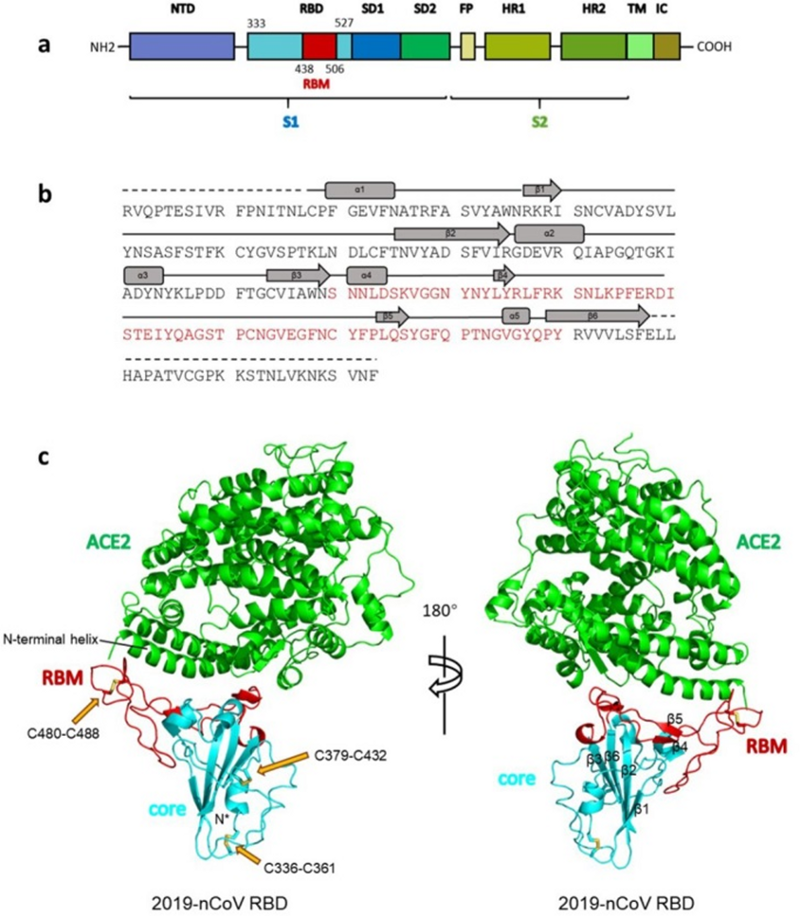
Общая структура RBD CoV2, связанного с ACE2.
(а) Общая топология мономера шиповидного белка 2019-nCov. NTD, N-терминальный домен. RBD, рецептор-связывающий домен. RBM, рецептор-связывающий мотив. SD1, поддомен 1. SD2, поддомен 2. FP, пептид слияния. HR1, гептадный повтор 1. HR2, гептадный повтор 2. TM, трансмембранная область. IC, внутриклеточный домен.
(б) Последовательность и вторичные структуры RBD 2019-nCov. RBM окрашен в красный цвет.
© Общая структура RBD 2019-nCov, связанного с ACE2. ACE2 окрашен в зеленый цвет. Ядро RBD 2019-nCoV окрашено в голубой цвет, а RBM — в красный. Дисульфидные связи в RBD 2019-nCov показаны в виде линии и обозначены желтыми стрелками. N-концевая спираль ACE2, ответственная за связывание, помечена.Исходный текстOverall structure of 2019-nCoV RBD bound with ACE2.
(a) Overall topology of 2019-nCoV spike monomer. NTD, N-terminal domain. RBD, receptor-binding domain. RBM, receptor-binding motif. SD1, subdomain 1. SD2, subdomain 2. FP, fusion peptide. HR1, heptad repeat 1. HR2, heptad repeat 2. TM, transmembrane region. IC, intracellular domain.
(b) Sequence and secondary structures of 2019-nCoV RBD. The RBM is colored red.
© Overall structure of 2019-nCoV RBD bound with ACE2. ACE2 is colored green. 2019-nCoV RBD core is colored cyan and RBM is colored red. Disulfide bonds in the 2019-nCoV RBD are shown as stick and indicated by yellow arrows. The N-terminal helix of ACE2 responsible for binding is labeled.
Так вот. Когда геном CoV2 только расшифровали, то сначала не было известно каких-то прям близкородственных ему штаммов. Но уже 23 января 2020 года Ши Чжэнли выпустила работу, в которой объявила, что CoV2 на 96% совпадает со штаммом RaTG13, который её лаборатория в 2013 году выделила из Юньнаньских летучих мышей. Правда, вне её лаборатории до января 2020 года об этом штамме не было известно никому.
Было сразу понятно, что RaTG13 — мальчик особенный. Взгляните на график:
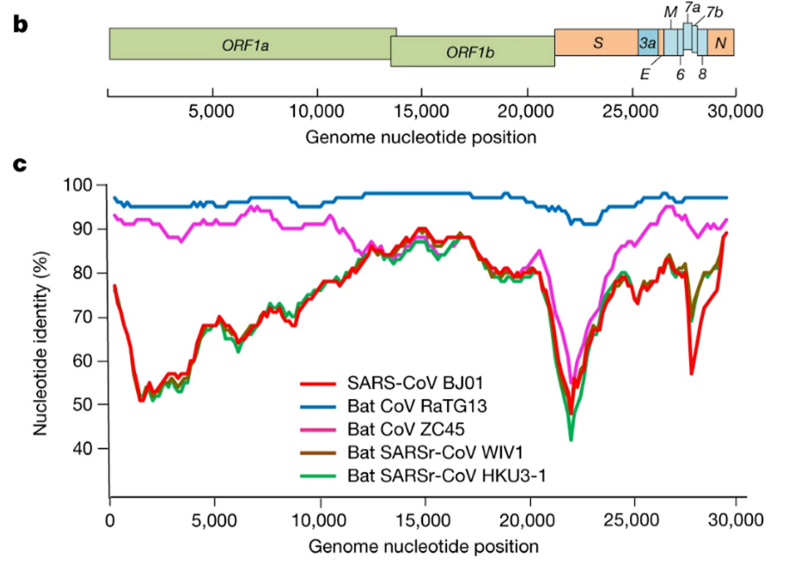
Это график схожести CoV2 с другими известными штаммами. Чем выше кривая, тем выше процент совпадения нуклеотидов. Как видим, в районе гена того самого шиповидного белка (S) только RaTG13 более-менее близок к CoV2, а все остальные штаммы в этом месте уходят в пике — как штаммы вирусов из других летучих мышей, так и первый SARS-CoV (красная кривая). Но тут пока нет ничего подозрительного — мало ли ещё неизвестных науке штаммов таят в себе неизведанные Юньнаньские пещеры? Ну да, не очень понятно как именно вирус оттуда добрался до Уханя, но чего не бывает.
Панголины
Далее на сцене появляются панголины: в феврале другая группа китайских учёных обнаружила в своих закромах штамм панголиньего коронавируса, который, хоть в целом и хуже чем RaTG13 был похож на CoV2 (на 90%), в том самом RBM шиповидного белка был почти идентичен — отличался лишь на 1 аминокислоту (см. два верхних сиквенса, точки означают совпадение с первым сиквенсом):
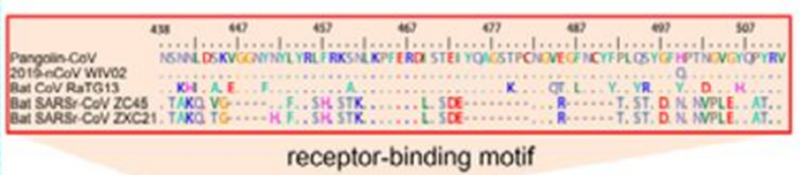
При этом в первой четверти S белка панголиний штамм на CoV2 не похож, а последовательность после RBM у всех троих штаммов (CoV2, Pangolin, RaTG13) более-менее совпадает. Но вот сам RBM у RaTG13 весьма отличается от CoV2, что видно по крутому провалу зелёного графика RaTG13 по сравнению с красным графиком CoV2 в районе RBM (вертикальная розовая полоса) на следующем графике:
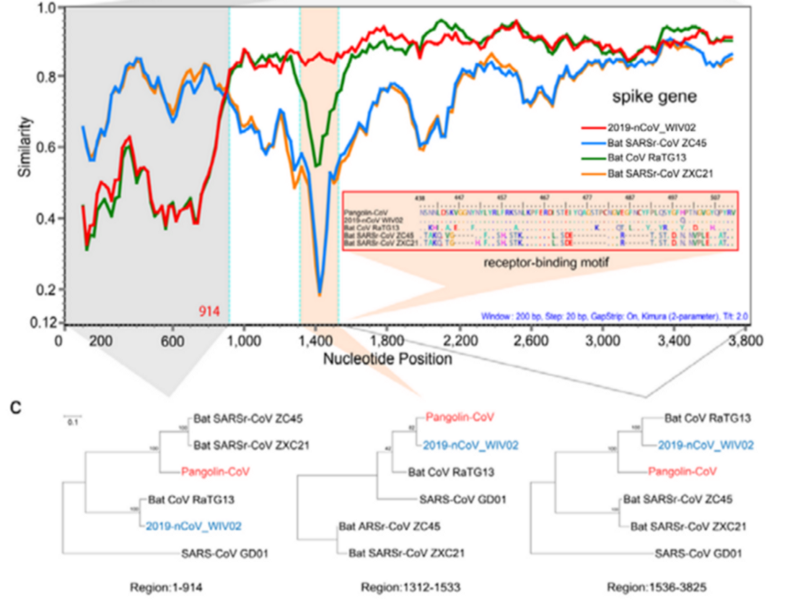
Эту разницу подтверждает и филогенетический анализ этих трёх областей, выделенных на графике выше — по RBM штамм панголина ближе к CoV2, чем RaTG13, а вот слева и справа от RBM к CoV2 ближе именно RaTG13. То есть налицо явная рекомбинация, о чем упоминают и сами авторы, и некоторые другие работы.
Кстати, откуда взялись у исследователей эти самые панголины? А вот отсюда:

Их конфисковала у контрабандистов китайская таможня и передала в реабилитационный центр в Гуандонге, где они скончались при выраженной коронавирусной симптоматике. Это, конечно, не могло не заинтересовать местных вирусологов, которые и выделили у них различный биоматериал:
Панголины, использованные в исследовании, были конфискованы таможней и Департаментом лесного хозяйства провинции Гуандун в марте-декабре 2019 года. В их число входят четыре китайских панголина (Manis pentadactyla) и 25 малайских панголинов (Manis javanica). Эти животные были отправлены в центр спасения диких животных; в основном они были неактивны и рыдали, и в конечном итоге умерли, несмотря на обширные усилия по их спасению. Образцы тканей были взяты из легких, лимфатических узлов, печени, селезенки, мышц, почек и других тканей у только что умерших панголинов, для гистопатологических и вирусологических исследований.Исходный текстPangolins used in the study were confiscated by Customs and Department of Forestry of Guangdong Province in March-December 2019. They include four Chinese pangolins (Manis pentadactyla) and 25 Malayan pangolins (Manis javanica). These animals were sent to the wildlife rescue center, and were mostly inactive and sobbing, and eventually died in custody despite exhausting rescue efforts. Tissue samples were taken from the lung, lymph nodes, liver, spleen, muscle, kidney, and other tissues from pangolins that had just died for histopathological and virological examinations.
Кстати, и не только местных, потому что другие китайские исследователи (гонконгские в данном случае) тоже получали образцы конфискованных панголинов и в феврале 2020 тоже выпустили схожую работу, отметив явные признаки рекомбинации в шиповидном белке CoV2:
Мы получили образцы замороженных тканей (легких, кишечника, крови), которые были взяты у 18 малайских панголинов (Manis javanica) в течение августа 2017 года — января 2018 года. Эти панголины были получены в ходе операций по борьбе с контрабандой таможней Гуанси. Поразительно, что высокопроизводительное секвенирование их РНК выявило присутствие коронавирусов в шести (два легких, два кишечника, одна смесь легких и кишечника, одна кровь) из 43 образцов.
…
Более заметным, однако, было наблюдение предполагаемых сигналов рекомбинации между коронавирусами панголинов, коронавирусами летучих мышей RaTG13 и 2019-CoV2 человека (Рис. 1c, d). В частности, 2019-CoV2 демонстрирует очень высокое сходство последовательностей с коронавирусами панголина из провинции Гуандун в рецептор-связывающем домене (RBD; сходство аминокислот 97,4%; обозначено красной стрелкой на рис. 1c и рис. 2a), хотя в оставшейся части вирусного генома 2019-CoV2 ближе всего к RaTG13. В то же время RaTG13 и 2019-CoV2 имеют только 89,2% сходства аминокислот в RBD. Коронавирусы панголинов из Гуандуна и 2019-CoV2 содержат идентичные аминокислоты в пяти критических остатках RBD, тогда как RaTG13 имеет только одну общую аминокислоту с 2019-CoV2 из этих пяти (остаток 442, нумерация SARS-CoV человека).Исходный текстWe received frozen tissue (lungs, intestine, blood) samples that were collected from 18 Malayan pangolins (Manis javanica) during August 2017-January 2018. These pangolins were obtained during the anti-smuggling operations by Guangxi Customs. Strikingly, high-throughput sequencing of their RNA revealed the presence of coronaviruses in six (two lung, two intestine, one lung-intestine mix, one blood) of 43 samples. With the sequence read data, and by filling gaps with amplicon sequencing, we were able to obtain six full or nearly full genome sequences — denoted GX/P1E, GX/P2V, GX/P3B, GX/P4L, GX/P5E and GX/P5L — that fall into the 2019-CoV2 lineage (within the genus Betacoronavirus) in a phylogenetic analysis (Figure 1a).
…
More notable, however, was the observation of putative recombination signals between the pangolins coronaviruses, bat coronaviruses RaTG13, and human 2019-CoV2 (Figure 1c, d). In particular, 2019-CoV2 exhibits very high sequence similarity to the Guangdong pangolin coronaviruses in the receptor-binding domain (RBD; 97.4% amino acid similarity; indicated by red arrow in Figure 1c and Figure 2a), even though it is most closely related to bat coronavirus RaTG13 in the remainder of the viral genome. Bat CoV RaTG and the human 2019-CoV2 have only 89.2% amino acid similarity in RBD. Indeed, the Guangdong pangolin coronaviruses and 2019-CoV2 possess identical amino acids at the five critical residues of the RBD, whereas RaTG13 only shares one amino acid with 2019-CoV2 (residue 442, human SARS-CoV numbering).
Кстати, авторы этой статьи тоже выделили явную филогенетическую мозаичность шиповидного белка CoV2:
Интересно, что филогенетический анализ только синонимичных сайтов в RBD показал, что филогенетическое положение гуандунского панголина согласуется с положением по остальной части вирусного генома, а именно, что он не является ближайшим родственником 2019-CoV2 (Figure 2b). Следовательно, возможно, что сходство аминокислот в RBD коронавирусов панголина и 2019-CoV2 обусловлено селективно-опосредованной конвергентной эволюцией, а не рекомбинацией, хотя на имеющихся данных трудно выбрать между этими сценариями.Исходный текстInterestingly, a phylogenetic analysis of synonymous sites alone in the RBD revealed that the phylogenetic position of the Guangdong pangolin is consistent with that in the remainder of the viral genome, rather than being the closest relative of 2019-CoV2 (Figure 2b). Hence, it is possible that the amino acid similarity between the RBD of the Guangdong pangolin coronaviruses and 2019-CoV2 is due to selectively-mediated convergent evolution rather than recombination, although it is difficult to choose between these scenarios on current data.
В переводе с научного, слова авторов означают, что если проанализировать весь RBD трёх штаммов, отбросив явные различия (несинонимичные замены) между ними, которые, в основном, приходятся на RBM (который, напомню, идентичен между CoV2 и Pangolin), и построить филогенетическое древо по синонимичным заменам, то CoV2 таки ближе к RaTG13, а не к панголиньему штамму. Что довольно странно в свете того, что у панголиньего с CoV2 идентичный RBM (то есть отрезок внутри RBD).
Далее авторы теоретизируют, что это может быть следствием конвергентной эволюции, то есть, иначе говоря, что штаммы CoV2 и панголинов пришли к идентичному RBM каждый своей дорогой, а не через совместную рекомбинацию общих предков. Потому что действительно уж очень странная должна была произойти рекомбинация — как будто кто-то просто взял кусок RBM из панголиньего штамма и заменил им RBM в RaTG13. А это уже смахивает не на эволюцию, а на, прости Дарвин, Intelligent Design.
Генеалогия коронованных особ
Для того чтобы получше понять происхождение CoV2, давайте ещё раз взглянем на последовательности шиповидного белка у нашей троицы: CoV2, RaTG13 и панголина — сравним попарную разницу между ними (точками отмечены идентичные аминокислоты, красные буквы показывают разницу, а прочерки обозначают удалённые/добавленные аминокислоты):

Невооружённым глазом видно, что в первой четверти сиквенса панголиний штамм далёк от CoV2 и RaTG13. Ну, а RaTG13, если бы не участок в районе RBM (красный прямоугольник), был бы ооочень близок к CoV2. Но, как я уже говорил, тот самый участок у CoV2 ближе всего к панголиньему штамму.
Кстати, а что там у других панголиньих штаммов? Давайте посмотрим. Ведь пока что мы анализировали только вирус, выделенный из панголинов, конфискованных таможней в 2019 году. А ещё была партия панголинов, конфискованных в 2017-ом, и у них тоже был выделен похожий штамм. Если сравнить RaTG13 с геномами вирусов из панголинов 2017 и 2019 годов, то тут тоже всё интересно:

В первой четверти S белка панголиньи штаммы от 2017 года ближе к RaTG13 (и CoV2) чем их панголиний собрат от 2019-го (MP789). При этом, у всех трёх явный недавний общий предок в областях, выделенных зелёными прямоугольниками, и в этих областях RaTG13 и панголин-19 (MP789) к нему ближе, чем панголин-17, так как у него с RaTG13 несколько общих мутаций (обведены красными и синими эллипсами), которых не наблюдается у легенды-17. При этом RBM у всех трёх разный, и разный примерно в одинаковой пропорции, и в схожих местах.
Возможно, уже после того как предки RaTG13 и MP789 разошлись, у MP789 произошла замена большого участка в первой четверти S белка (которой не произошло у RaTG13 и панголина-17), а остальная часть S белка у всех троих осталась общей. А потом пути генофондов RaTG13 и MP789 сошлись опять и в порыве страсти породили CoV2. Также вполне возможно, что предок RaTG13 является результатом рекомбинации предков панголиньих штаммов.
Ещё любопытно видеть довольно уникальную одинаковую мутацию (QTQTNS) у RaTG13 и панголина-19 прямо перед тем местом, где у CoV2 находится новый furin cleavage site, возникший из-за врезки 4-х новых аминокислот в этом месте (PRRA). Если посмотреть на нуклеотидную последовательность вокруг этой одинаковой мутации, то видно, что RaTG13 и CoV2 по ней ближе друг к другу, чем к панголину-19, так как успели накопить несколько общих мутаций (выделены синим):

Кстати, с Orf1ab у CoV2 тоже филогенетическая чехарда: 1a ближе к RaTG13, а вот 1b — к панголину-19:
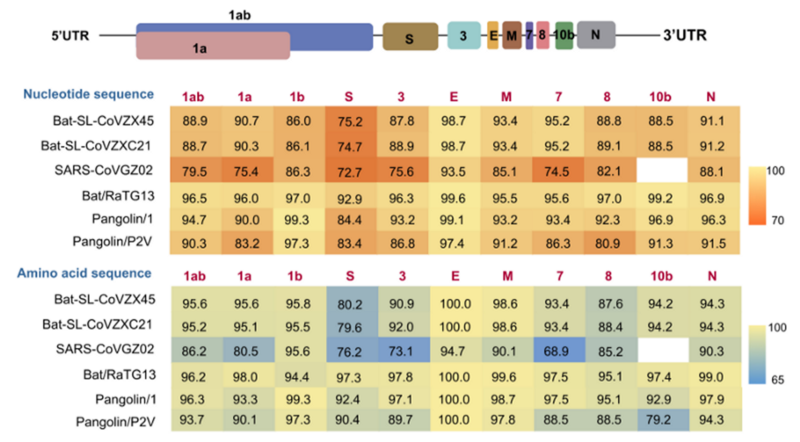
То есть получается, что предок CoV2 согрешил с общим предком панголина-19, как минимум, дважды? Впервые — когда он (вместе с общим предком с RaTG13) унаследовал Orf1ab и вторую часть шиповидного белка с QTQTNS мутацией, а вторично — когда приобрёл 1b вместе с RBM, уже отличающимся от RaTG13-шных. Нет, конечно, такое возможно и само по себе не особо примечательно — ведь эти вирусы мутируют и рекомбинируют постоянно. Другой вопрос, где именно летучемышиные и панголиньи вирусы могут друг с другом встречаться для таких оргий — в горных пещерах, «мокрых рынках», приютах для конфискованных животных, или даже в лабораториях. Но давайте повременим с этими вопросами. Сначала рассмотрим самый бросающийся в глаза аспект нового вируса — врезку из 4-х аминокислот, превратившую его в супер-убийцу.
Врежу аккуратно, но сильно
Совершенно невозможно игнорировать эту врезку PRRA между S1 и S2. Как заноза торчит она в геноме нашего нового CoV2. Врезка эта не простая, а золотая. Она создаёт тот самый furin cleavage site, о котором я упомянул в самом начале. Что это за зверь? Попытаюсь вкратце объяснить. Помните структуру нашего шиповидного белка? Вот наглядная диаграмма:
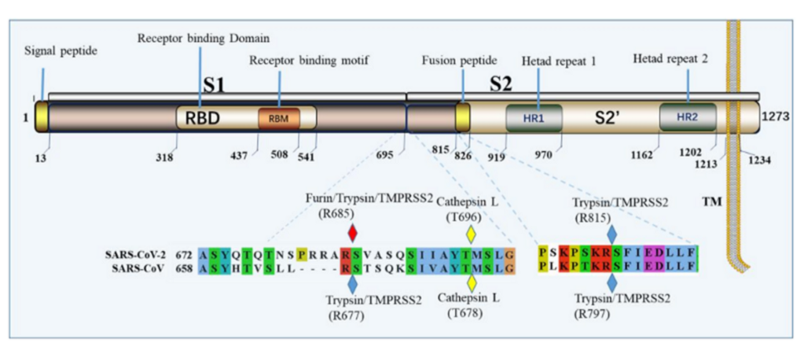
Белок состоит из двух частей, S1 и S2, из которых S1 отвечает за первичный контакт с рецептором (тот самый Receptor Binding Domain/Motif), а S2 отвечает уже за слияние с клеточной мембраной и проникновение в клетку. Запускает процесс слияния отмеченный жёлтым fusion peptide, но для того чтобы он начал своё грязное дело, кто-то должен разрезать S белок в одном из сайтов, выделенных ромбиками на диаграмме выше. Своих таких «резальщиков» у вируса нет (есть другие, но это к делу не относится), поэтому он полагается на различные протеазы своих жертв, благо таких протеаз, как можно понять по обилию цветов тех самых ромбиков, существует несколько видов. Но не все они равны, и не во всех типах клеток есть нужные вирусу протеазы. А фурин как раз один из самых эффективных, да ещё и обитает он не только на поверхности клеток, но и внутри. Нагляднее всего опасность нового фуринового сайта демонстрирует разница между CoV2 и его «предтечей» SARS-CoV:
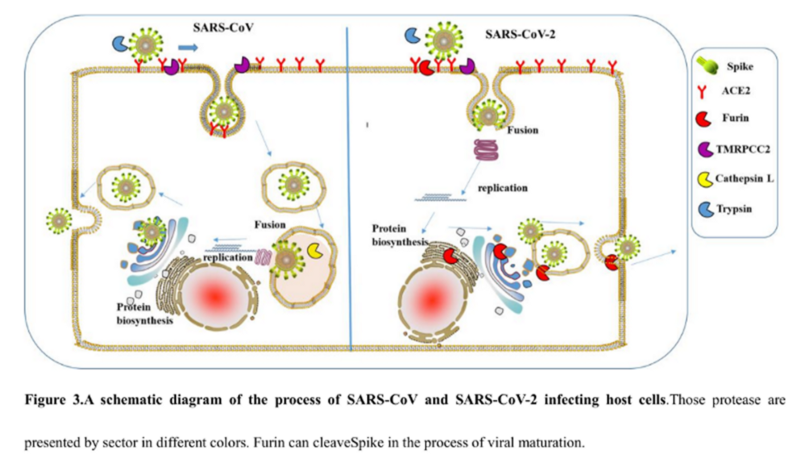
Как видно из диаграммы, в случае CoV2, благодаря фуриновому сайту, разрезать его S белок вне клетки могут не два, а три класса протеаз (три разноцветных пакмэна). Но, быть может, самая важная разница состоит в том, что фурин присутствует и внутри клетки, поэтому он может разрезать S белок сразу после сборки вириона, тем самым повышая способность новых вирионов сливаться с другими клетками — так сказать, не отходя от кассы.
Кстати, существует вероятность, что именно новый фуриновый сайт играет важную роль в выраженной возрастозависимой морбидности и летальности CoV2:
По неизвестным причинам, пациенты с гипертонией, диабетом, ишемической болезнью сердца, цереброваскулярными заболеваниями, хронической обструктивной болезнью легких и почечной дисфункцией имеют худшие клинические исходы при инфицировании SARS-CoV-2. Целью данного обзора является обобщение доказательств существования повышенного плазмин (оген)а у пациентов с COVID-19 с этими коморбидными состояниями. Плазмин и другие протеазы могут расщеплять новый фуриновый сайт в шиповидном белке SARS-CoV-2 внеклеточно, что увеличивает его инфекционность и вирулентность.Исходный текстPatients with hypertension, diabetes, coronary heart disease, cerebrovascular illness, chronic obstructive pulmonary disease, and kidney dysfunction have worse clinical outcomes when infected with SARS-CoV-2, for unknown reasons. The purpose of this review is to summarize the evidence for the existence of elevated plasmin (ogen) in COVID-19 patients with these comorbid conditions. Plasmin, and other proteases, may cleave a newly inserted furin site in the S protein of SARS-CoV-2, extracellularly, which increases its infectivity and virulence.
Ах, да, разрезает фурин белки в строго определённых местах, а именно после последовательности RxxR (то есть Arg-X-X-Arg, где X может быть любой аминокислотой). При этом если на втором или третьем месте тоже аргинин (то есть RRxR или RxRR), то эффективность расщепления этого сайта существенно возрастает.
Поэтому появление нового furin cleavage site специалистами было замечено сразу. Ещё бы! Ведь такого сайта нет ни у кого из ближайших или даже дальних родственников Cov2 — те коронавирусы, у которых он есть, лишь на 40% близки к Cov2 по геному:
Было обнаружено, что все шиповидные белки с гомологией белку SARS-CoV-2, превышающей 40%, не имели сайта расщепления фурином (рисунок 1, таблица 1), включая Bat-CoV RaTG13 и SARS-CoV (с идентичностью последовательности как 97,4% и 78,6% соответственно). Фуриновый сайт «RRAR» в SARS-CoV-2 является уникальным в своем семействе, благодаря уникальной вставке «PRRA». Этот сайт в SARS-CoV-2 вряд ли мог эволюционировать из MERS, HCoV-HKU1 и т.д. Из имеющихся в настоящее время последовательностей в базах данных нам трудно найти источник. Возможно, есть еще много эволюционных промежуточных последовательностей, ожидающих открытия.Исходный текстIt was found that all Spike with a SARS-CoV-2 Spike sequence homology greater than 40% did not have a furin cleavage site (Figure 1, Table 1), including Bat-CoV RaTG13 and SARS-CoV (with sequence identity as 97.4% and 78.6%, respectively). The furin cleavage site «RRAR» in SARS-CoV-2 is unique in its family, rendering by its unique insert of «PRRA». The furin cleavage site of SARS-CoV-2 is unlikely to have evolved from MERS, HCoV-HKU1, and so on. From the currently available sequences in databases, it is difficult for us to find the source. Perhaps there are still many evolutionary intermediate sequences waiting to be discovered.
Вот наглядная иллюстрация из той же статьи, что и вышеприведённая цитата (розовым отмечены коронавирусы с фуриновым сайтом, на 10 часах приведены 3 разных штамма Cov2):

Ближайший родственник с фуриновым сайтом — это штамм HKU5, выделенный командой Ши Чжэнли в 2014 году в Гуанчжоу из летучих мышей рода Pipistrellus (добавлен в GenBank в 2018-ом). Но родственник он весьма дальний — их шиповидные белки совпадают лишь на 36%.
В общем, учёные в замешательстве. Откуда взялась эта вставка из 12 нуклеотидов? Может ли она быть рукотворной? Вполне. Ведь такими вставками вирусологи занимались многократно, причём издавна. Например, американцы вставляли RRSRR в шиповидный белок первого SARS-CoV ещё в 2006-ом:
Чтобы исследовать, может ли протеолитическое расщепление в месте последовательсти основных аминокислотных остатков способствовать клеточной активности слияния, мы мутировали шиповидный белок SARS-CoV, чтобы создать сайт узнавания фурином (RRSRR) в одном из двух мест.Исходный текстTo investigate whether proteolytic cleavage at the basic amino acid residues, were it to occur, might facilitate cell–cell fusion activity, we mutated the wild-type SARS-CoV glycoprotein to construct a prototypic furin recognition site (RRSRR) at either position.

А японцы вставили такой сайт (RRKR) в белок SARS-CoV в 2008 году, правда немного ниже по течению:
В том же 2008-ом году их голландские коллеги тоже изучали эти протеазные сайты у SARS-CoV и сравнивали их с мышиным коронавирусом MHV, у которого этот сайт есть (SRRAHR|SV), причём похожий на сайт нашего CoV2 (SPRRAR|SV):
В 2009 году другая американская группа тоже решила поупражняться над «улучшением» SARS-CoV и, не изменяя американской традиции не мелочиться с аргининами, они вставили RRSRR:
Чтобы исследовать потенциальное использование SARS-CoV S1-S2 и S2 «положений в качестве сайтов для протеолитического расщепления, мы сначала ввели сайты распознавания расщепления фурином в этих местах, сделав следующие мутации 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1-S2) и 792-LKPTKRSF-LKRTKRSF-799 (S2 »).Исходный текстTo examine the potential use of the SARS-CoV S1–S2 and S2? positions as sites for proteolytic cleavage, we first introduced furin cleavage recognition sites at these locations by making the following mutations 664-SLLRSTSQSI — SLLRRSRRSI-671 (S1–S2) and 792-LKPTKRSF — LKRTKRSF-799 (S2?).
Пекин-2019
Но самой недавней подобной работой, которую я видел, была работа октября 2019 года учёных из Пекина, где новый фуриновый сайт RRKR вставили не в какой-то псевдовирус, а в самый настоящий куриный коронавирус, infectious bronchitis virus (IBV), что привело к ~100-кратному увеличению вирусных титров у синтетического мутанта (панель B ниже):
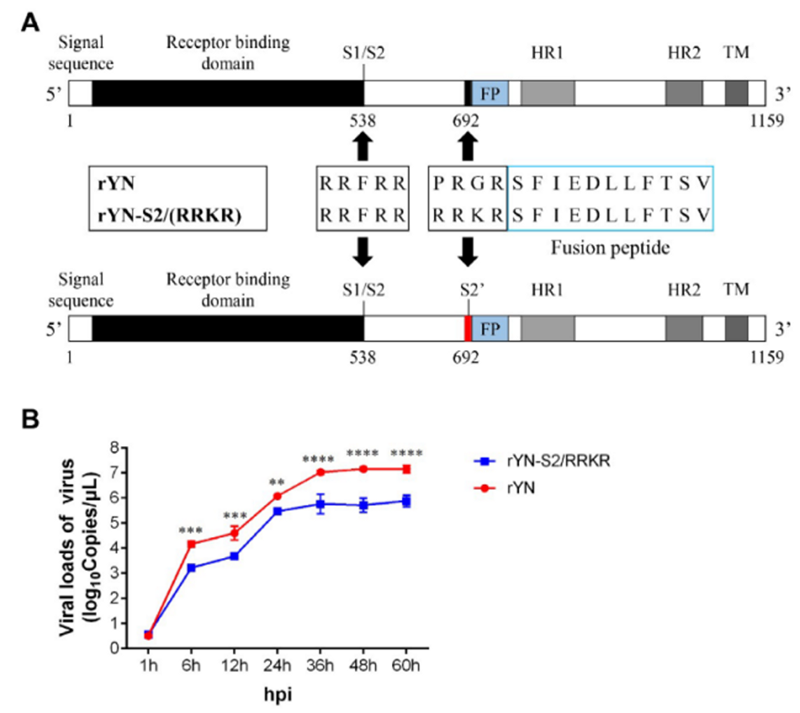
Кстати, интересно упоминание авторов, что добавление фуринового сайта позволяет вирусу-мутанту поражать нервные клетки. Быть может, именно фуриновый сайт CoV2 является причиной того, что некоторые пациенты с CoV2 демонстрируют неврологическую симптоматику, включая потерю обоняния:
Мутация сайта S2' шиповидного белка рекомбинантного вируса генотипа QX приводит к более высокой патогенности, выраженным нервным симптомам и нейротропизму по сравнению у цыплят, по сравнению с цыплятами, инфицированными вурусом дикого типа IBV (WT-IBV). В этом исследовании мы представляем доказательства того, что рекомбинантный IBV с мутантным сайтом S2 (фуриновый сайт-S2) приводит к более высокой смертности. Заражение мутантом IBV вызывает тяжелый энцефалит и разрушает гематоэнцефалический барьер.
…
Таким образом, наши результаты демонстрируют, что фуриновый сайт перед FP в шиповидном белке является важным сайтом для CoV, модулирующим проникновение, слияние клеток с вирусом, адаптацию к клетке-хозяину, клеточный тропизм и патогенность, но не антигенность.Исходный текстMutation of the S2' site of QX genotype (QX-type) spike protein (S) in a recombinant virus background results in higher pathogenicity, pronounced neural symptoms and neurotropism when compared with conditions in wild-type IBV (WT-IBV) infected chickens. In this study, we present evidence suggesting that recombinant IBV with a mutant S2' site (furin-S2' site) leads to higher mortality. Infection with mutant IBV induces severe encephalitis and breaks the blood–brain barrier.
…
In summary, our results demonstrate that the furin cleavage site upstream of the FP in S protein is an important site for CoV, modulating entry, cell–virus fusion, adaptation to its host cell, cell tropism and pathogenicity, but not antigenicity.
При этом у многих коронавирусов фуриновые сайты, конечно же, встречаются в природе, и они весьма разнообразны. Да и появляться они могут и в результате случайных мутаций. Именно это произошло в случае с MERS-ом, о чем в 2015 нам поведал международный коллектив авторов, среди которых были и Ши Чжэнли, и Ральф Барик — две звезды синтетической коронавирусологии. О них мы ещё неоднократно вспомним, а пока пару слов о той статье. Собственно в ней авторы показали две ключевые мутации, которые позволили MERS перескочить с летучих мышей не человека. И одна из этих мутаций привела к возникновению фуринового сайта. Правда это была не врезка новых аминокислот, а мутации ранее существующих (отмеченных красным ниже):
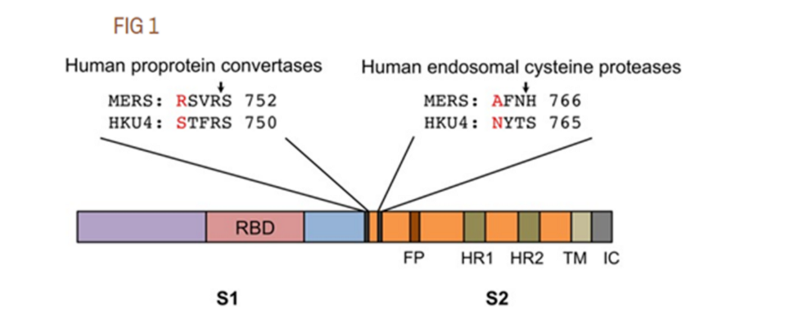
Авторы не просто показали, а привязали эти мутации обратно к исходному летучемышиному шиповидному белку, создав в нём те же самые мутации (то есть, и фуриновый сайт), и показав, что это придаёт ему способность заражать человеческие клетки:
Чтобы оценить потенциальные генетические изменения, необходимые для HKU4 для заражения клеток человека, мы изменили шиповидный белок HKU4, стремясь повысить его способность опосредовать проникновение вируса в клетки человека. С этой целью мы ввели две одиночные мутации, S746R и N762A, в белок HKU4.
…
Следовательно, реинжинирированные мотивы hPPC и hECP позволили активировать спайк HKU4 человеческими эндогенными протеазами и тем самым позволили псевдовирусам HKU4 обойти необходимость проникновения экзогенных протеаз в клетки человека. Эти результаты показывают, что спайку HKU4 нужны только две одиночные мутации на границе S1/S2, чтобы получить полную способность опосредовать проникновение вируса в клетки человека.Исходный текстTo evaluate the potential genetic changes required for HKU4 to infect human cells, we reengineered HKU4 spike, aiming to build its capacity to mediate viral entry into human cells. To this end, we introduced two single mutations, S746R and N762A, into HKU4 spike. The S746R mutation was expected to restore the hPPC motif in HKU4 spike, whereas the N762A mutation likely disrupted the potential N-linked glycosylation site in the hECP motif in HKU4 spike.
…
Therefore, the reengineered hPPC and hECP motifs enabled HKU4 spike to be activated by human endogenous proteases and thereby allowed HKU4 pseudoviruses to bypass the need for exogenous proteases to enter human cells. These results reveal that HKU4 spike needs only two single mutations at the S1/S2 boundary to gain the full capacity to mediate viral entry into human cells.
Кстати, то как они это сделали может людей далёких от современных биотехнологий напугать само по себе — потому что авторы вставили этот коронавирусный шиповидный белок в инактивированный ретровирус (ВИЧ):
Вкратце, псевдотипированные ретровирусы MERS-CoV-spike, экспрессирующие репортерный ген люциферазы, были получены путем котрансфекции клеток HEK293T плазмидой, несущей геном ВИЧ-1, дефектный по Env, экспрессирующий люциферазу (pNL4–3.luc.RE-) и плазмидой, кодирующей MERS-CoV шиповидный белок.Исходный текстBriefly, MERS-CoV-spike-pseudotyped retroviruses expressing a luciferase reporter gene were prepared by cotransfecting HEK293T cells with a plasmid carrying Env-defective, luciferase-expressing HIV-1 genome (pNL4–3.luc.R-E-) and a plasmid encoding MERS-CoV spike protein.
Быть может, именно это подвигло индийских исследователей на поиск схожих у ВИЧ и CoV2 последовательностей в геноме (но их препринт быстро раскритиковали за неудачную методологию и ошибочные выводы). На самом деле такие псевдовирусы специалисты используют регулярно, и вообще, не стоит огульно бояться ретровирусов как класс — их подвид лентивирусы используется для генной терапии уже много лет.
Откуда взялся RaTG13
Вообще, RaTG13 — феноменальный штамм. Странно, что все эти годы группа Ши Чжэнли о нём молчала. Ведь он совсем не похож на остальных своих собратьев, особенно в последовательности шиповидного белка, а это, напомню, именно то место, которое определяет к каким типам клеток (и каких видов) данный вирус сможет цепляться. Вот график схожести генома CoV2 в сравнении с другими летучемышиными коронавирусами (панель B):
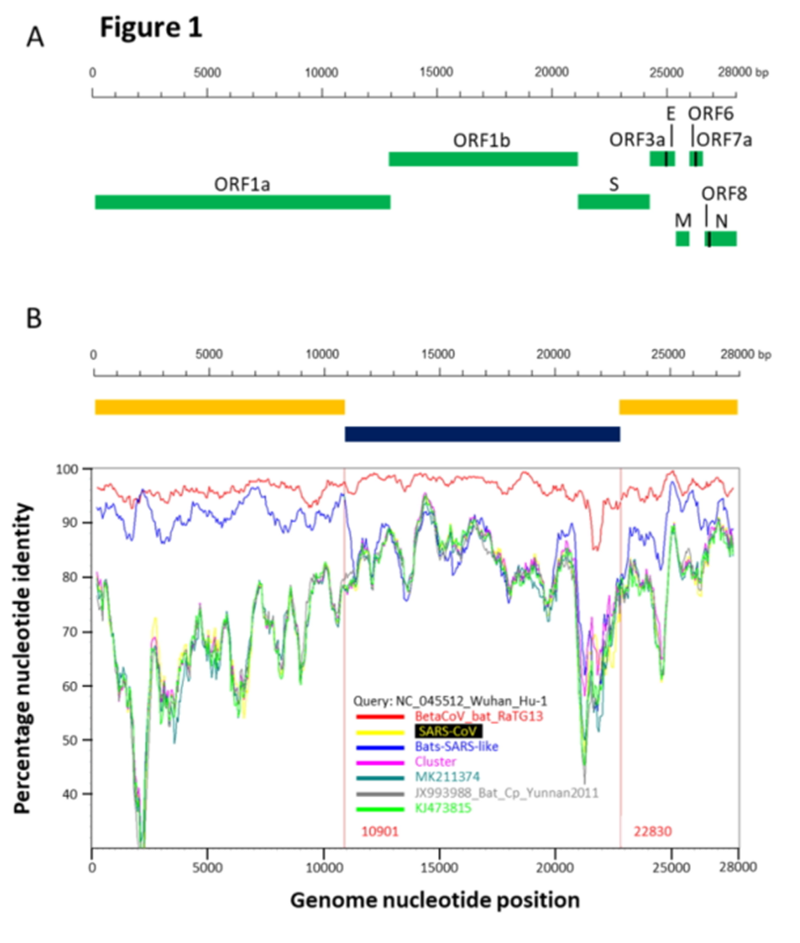
RaTG13 — это красная кривая, а синяя — это наиболее близкие к RaTG13 штаммы (ZXC21 и ZC45). Эти штаммы были выделены из Китайских подковоносов (Rhinolophus sinicus) в Чжоушане в 2015 (ZXC21) и 2017 (ZC45) годах. Как видно из графика, даже они очень сильно расходятся с RaTG13 (красная кривая) в районе S белка. При этом график выше не так хорошо передаёт масштаб пропасти между ними как прямое сравнение сиквенсов:

Как видим, шиповидные белки ZXC21 и ZC45 не только, в целом, на 23–24 аминокислотных остатка короче белка RaTG13, но они короче в самом важном месте — в RBM (см. делеции в красном прямоугольнике, отмеченные красными прочерками).
Так откуда же взялся RaTG13? Как я уже сказал, в 2020 году Ши Чжэнли сообщила, что выделила его у подковоносов (только вида Rhinolophus affinis, а не R. sinicus) в июле 2013 года в Юньнани. Правда, до конца января 2020 года публично о его существовании не сообщалось, а вот как описывает своё знаменательное открытие о его схожести с CoV2 сама группа Ши Чжэнли:
Затем мы обнаружили, что короткий участок РНК-зависимой РНК-полимеразы (RdRp) от коронавируса летучей мыши (BatCoV RaTG13), который ранее был обнаружен у Rhinolophus affinis из провинции Юньнань, показал высокую идентичность последовательности 2019-CoV2. Мы провели полноразмерное секвенирование на этом образце РНК (регистрационный номер GISAID EPI_ISL_402131). Анализ Simplot показал, что 2019-CoV2 очень похож на RaTG13 (Fig. 1c), с общей идентичностью генома 96,2%.Исходный текстWe then found that a short region of RNA-dependent RNA polymerase (RdRp) from a bat coronavirus (BatCoV RaTG13) — which was previously detected in Rhinolophus affinis from Yunnan province — showed high sequence identity to 2019-CoV2. We carried out full-length sequencing on this RNA sample (GISAID accession number EPI_ISL_402131). Simplot analysis showed that 2019-CoV2 was highly similar throughout the genome to RaTG13 (Fig. 1c), with an overall genome sequence identity of 96.2%.
Негусто: ну был у них этот штамм «ранее обнаружен», и был. Лежал на полке. До 2020 года сиквенировали в нём только часть генома, отвечающую за RdRp. Откуда он на полке взялся? В Юньнани в 2013 году выделили. Где именно? Не сказали. В GenBank тоже не было этой информации. Но, к счастью, в базе геномов GISAID была: сообщается, что в городе Пуэр (да, на родине любимого чая Басты) его выделили из fecal swab некого самца:

Это меня немного заинтриговало, потому что в моих блужданиях по Пабмеду, я уже натыкался на экспедицию в Пуэр летом 2013 года:
Летучие мыши были пойманы в различных местах в пяти округах четырех префектур провинции Юньнань, Китай, с мая по июль 2013 года.Исходный текстBats were captured from various locations in five counties of four prefectures of Yunnan Province, China, from May to July 2013.
В той экспедиции ничего особо интересного для нас исследователи не нашли, но, быть может, именно тогда Ши Чжэнли (или кто-то из её группы?) выделил тот самый образец RaTG13? Который они сиквенировали лишь частично, но почему-то реши
