Прионная болезнь. Романтический ужас
С прионными болезнями человечество знакомо давно. Например, в Англии ещё в 1732˗˗ 1755 годах была распространена болезнь овец под названием «скрепи». Долгое время получение точных данных об этом заболевании было затруднено, вследствие того что такие сведения нередко умышленно скрывали по коммерческим соображениям.
Тем не менее, постепенно скрепи быстро распространилась среди племенных овец. Этому способствовала практика инбридинга для улучшения качества шерсти. Поскольку позднее от этого метода ушли, случаи скрепи стали реже, но полностью не исчезли.
 Больная срепи овца
Больная срепи овца
Для обозначения скрепи используется более тридцати различных наименований. Только в Англии заболевание получило такие названия, как «вертячка», «почесуха», «трясучка» и даже «рахит». Существовали и другие названия, в которых так или иначе отражались характерные признаки клинического проявления болезни, связанные со стремлением животных чесаться. Примечательно, что одним из названий скрепи во Франции было «La tremblante», что в переводе значит «дрожать».
Несмотря на широкое распространение, еще не так давно скрепи рассматривали как чисто сельскохозяйственную проблему. Природа заболевания оставалась неизвестной, хотя более ста лет назад уже предпринимались первые попытки продемонстрировать заразность и возможность передачи этого заболевания. Но лишь в 1899 году впервые удалось передать скрепи здоровой овце путём искусственного заражения мозговой тканью, взятой от больного животного.
В начале ХХ века высказывались мнения о паразитарной этиологии скрепи, о возможной роли корнибактерий и других агентов. В 1926 году во Франции было предположено, что эту болезнь вызывает вирус, однако для реализации его инфекционного действия необходимы соответствующие условия в организме. И только лишь десять лет спустя учёные из Франции доказали, что возбудитель скрепи способен проходить через тончайшие бактериальные фильтры и заражать этим фильтратом здоровых овец.
Несмотря на широкое распространение, долгое время скрепи оставалась малоизвестной болезнью, о которой знали только ветеринары.
 Альфонс Мария Якоб 02.07.1884 — 17.10.1931
Альфонс Мария Якоб 02.07.1884 — 17.10.1931
В начале 20-х годов ХХ века, когда во Франции шли исследования скрепи, немецкий врач Альфонс Мария Якоб описал пять случаев необычного заболевания человека, отличавшегося своеобразными клиническими симптомами и необычными изменениями в ЦНС у людей в возрасте от тридцати до пятидесяти лет. Обнаруживаемые у пациентов изменения были сходны только с изменениями, описанными ранее Гансом Герхардом Кройтцфельдтом, обнаруженными им при исследовании тела двадцатилетней девушки по имени Берта Е. Несчастная Берта при жизни страдала расстройствами зрения, навязчивыми мыслями об одержимости, нарушением координации движений и эпилептическими припадками. Учитывая эти обстоятельства, было принято решение назвать новое заболевание болезнью Кройтцфельдта-Якоба. Справедливости ради стоит сказать, что изменения, которые обнаружили Кройтцфельдт и Якоб, уже были известны учёным.
 Ганс Герхард Кройтцфельд 02.06.1885 — 30.12.1964
Ганс Герхард Кройтцфельд 02.06.1885 — 30.12.1964
Именно такие изменения в ЦНС описывали ветеринары у овец, погибших от скрепи, но сообщения об этом прошли мимо врачебного сообщества.С 1920 по 1968 годы в мировой литературе было описано всего сто восемьдесят случаев, в то время как за последующее десятилетие — более двух тысяч. Однако русскоязычных источниках того периода можно обнаружить только четыре случая подобного заболевания. Резкое увеличение исследований за прошедшее время объясняется возможностью передачи этого заболевания от человека к человеку. Все эти случаи объединили в группу энцефалита неясного происхождения. Так это и продолжалось, пока с юго-западной части Тихого океана не стали приходить любопытные новости.
Собственно, открытие этой группы болезней началось с обычного наблюдения…
В восточной части острова Новая Гвинея на высоте 1500 — 2000 метров над уровнем моря расположен труднодоступный горный район. Здесь, к югу и востоку от горы Маклая, расположены туземные деревни языковых групп форе, кейагана, каните, ауйяна, кими и узуруфа (вскоре читателю станет ясно, какую роль сыграют языковые различия в течении эпидемических процессов). Согласно древним обычаям, между племенами и деревнями велись частые и кровопролитные войны с ритуальными убийствами, колдовством и людоедством.
 Аборигены форе
Аборигены форе
В 1949 году в Тарибо (область Кейагана) была организована первая лютеранская миссия, а вскоре и первый патрульный пост, созданный специально для прекращения междоусобицы и поддержания порядка. Первый патрульный офицер района Джон МакАртур 6 декабря 1953 года, проходя к деревне Амуси, увидел сидящую у костра маленькую девочку. Джон МакАртур писал в своём дневнике:
»Она сильно дрожала, а голова её покачивалась из стороны в сторону. Мне сказали, что она жертва колдовства и что эта дрожь продолжится вплоть до её смерти. До самой смерти она не сможет есть. Через несколько недель она должна погибнуть».[1] |
 Ребенок с прогрессирующей курой. Он не может
ходить или сидеть прямо без посторонней
помощи и серьезно не доедает.
Источник: https://ru.abcdef.wiki/wiki/Kuru_(disease)
Ребенок с прогрессирующей курой. Он не может
ходить или сидеть прямо без посторонней
помощи и серьезно не доедает.
Источник: https://ru.abcdef.wiki/wiki/Kuru_(disease)
Джон МакАртур продолжал встречать среди туземцев подобные случаи заболевания, которое, как он вскоре установил, называлось «куру» ˗˗ словом, означающим у папуасов Новой Гвинеи дрожание от холода или от страха. Более того он оказался достаточно проницательным человеком, предположив, что куру — новое, неизвестное в то время заболевание человека. Эти данные, в виде короткого сообщения получил Австралийский врач прибалтийского происхождения Винсент Зигас. В результате с 1959 года он начал наблюдения над больными аборигенами. В октябре того же года Зигас провёл несколько недель в области Окапа, где собрал сыворотки крови у двадцати шести больных куру и даже сумел взять некроптаты мозга человека, умершего от этого заболевания. Но первые попытки лабораторного исследования заболевания не внесли ясности в природу куру.
В начале марта 1957 года в изучение куру включился американский вирусолог словако-венгерского происхождения Карлтон Гайдушек. После этого началось интенсивное и плодотворное исследование неизвестной ранее болезни. Гайдушек был педиатром по образованию и вирусологом по призванию.
 Даниел Карлтон Гайдушек 09.09.1923 — 12.12.2008
Даниел Карлтон Гайдушек 09.09.1923 — 12.12.2008
Значительную помощь в исследовании нового заболевания оказывал невропатолог из Национального института нервных болезней США Игорь Клатцо, на его плечи легла вся техническая сторона вопроса. Именно Клатцо обратил внимание, что увиденная им картина изменений мозга напоминает гистологический препарат, который он изучал в коллекции своего учителя Оскара Фогта. Картинка была столь необычной и запоминающейся, что Клатцо решил отправить сообщение на ежегодную медицинскую выставку в Лондон. После окончания выставки Клатцо получил сообщение от английского ветеринара Хаддлоу, который указывал на сходство патологических изменений в мозгу овец, погибших от скрепи, и опубликованных препаратов, полученных от пациентов их Новой Гвинеи.
Хадлоу сравнил в своей работе скрепи и куру и пришёл к выводу, что эти заболевания родственны между собой. С этого момента скрепи стала привлекать к себе всё более пристальное внимание со стороны врачей.
Картина начала складываться.
Несмотря на энергичные меры, прекрасную организацию исследований и их комплексный характер, выяснение причин куру или, точнее, инфекционной природы этого заболевания потребовало ещё девять лет напряжённого труда. Однако значительно раньше, в 1957 году, в двух опубликованных работах уже были описаны клиника куру и особенности течения этой болезни. В 1963 году, для того чтобы подтвердить предположение об инфекционной природе заболевания, Гайдушек и Гиббс ввели в мозг самке и самцу шимпанзе суспензию мозговой ткани двух погибших от куру людей. Самка заболела спустя двадцать один месяц, самец — через тридцать месяцев после введения исследуемого материала. И клинические признаки медленно прогрессирующего заболевания, и картина патогистологических изменений только в ЦНС заражённых и погибших животных были практически неотличимы от таковых у заболевших и погибших от куру людей.
Эти данные вскоре были подтверждены возможностью серийного пассирования материала и передачи таким путём заболевания от шимпанзе к шимпанзе. Эти опыты дали возможность накопить немало ценных сведений, сразу же позволивших говорить о возбудителе куру. Именно в ходе этих экспериментов было установлено, что возбудитель куру проходит через миллипоровые фильтры с диаметром пор 100 нм. |
В суспензии мозговой ткани, приготовленной на фосфатно-буферном растворе, материал сохраняет инфекционность при -70° С в течение пяти лет. Возбудитель необычайно термоустойчив: тридцатиминутное прогревание при 85°С не разрушает и заметно не снижает его инфекционности. Более того, даже кипячение в течение тридцати минут не инактивирует полностью инфекционный материал. Возбудитель устойчив к обработке формальдегидом, глутаральдегидом, в-пропионлактоном, ЭДТА, рибонуклеазой А и III, дезоксирибонуклеазой I. Совершенно никакого инактивирующего воздействия не оказывал на инфекционного агента ультразвук, ионизирующая радиация и УФ-дучи. Этот агент оказался во всех смыслах «твёрдым орешком» и задал множество загадок исследователям.
Первоначально прионные болезни имели ряд особенностей, которые наводили на мысль об их генетической природе, поскольку куру встречалась в единственном месте на Земле, то есть являлась эндемичным заболеванием. Его изолированность природными условиями, естественно, вызывала представление о формировании генетически контролируемой восприимчивости к куру. Первые же исследователи этого заболевания рассматривали куру как наследственно-семейное дегенеративное заболевание ЦНС. Представление о генетической детерминированности куру опирались на ряд эпидемиологических наблюдений:
Высокая заболеваемость народности лингвистической группы форе, достигавшая 30,8 на 1000 жителей, в то время как заболеваемость в племенах людоедов, расположенных в непосредственном соседстве, оказывалась значительно меньшей или отсутствовала. К этому следует добавить случаи заболевания у представителей народности форе, ранее покинувших эндемичный район, и отсутствие заболевания среди обслуживающего персонала, побывавшего для оказания медицинской помощи из других регионов Новой Гвинеи или других стран мира.
Долгое время казалось вполне обоснованным представление о существовании у жителей эндемичного региона аутосомального гена К (от названия заболевания «куру»), детерминирующего восприимчивость к этой болезни, доминантного у женщин и рецессивного у мужчин.
Предрасположенным к куру считались гомозиготные индивидуумы обоего пола и гетерозиготные взрослые женщины. К этому следует добавить выявленную связь восприимчивости к инфекционному агенту с определенными группоспецифичными белками сыворотки крови. Однако упомянутые выше данные о возбудителе куру, случаи заболевания куру среди женщин из соседних племён, вступивших в брак с мужчинами народности форе, а также необычайно высокая частота появления куру среди этой народности, значительно превышающая частоту мутации любого известного гена, ˗˗ все эти данные не позволили признать существование генетической предрасположенности к куру.
Следует подчеркнуть, что в те времена также считалось, что этиологическим фактором медленных инфекций является вирус, однако основным возражением против этого предположения была как раз-таки высокая резистенстность возбудителя к УФ-лучам и ионизирующей радиации. Позже английские исследователи впервые продемонстрировали, что для инактивации возбудителя скрепи в мозговых гомогенатах необходимы очень высокие дозы радиации. На основании этого наблюдения они пришли к выводам, что размеры возбудителя прионных болезней меньше размеров вируса, а в его составе нет нуклеиновых кислот.
Эти данные послужили основанием для ряда концепций, главная из которых — белковая гипотеза, согласно которой возбудитель медленных инфекций представляет собой самореплицирующуюся белковую молекулу.
Для того чтобы поставить последнюю точку в вопросе групповой принадлежности возбудителей медленных инфекций, потребовалось решить задачу высокой степени очистки. В 1980 году Гайдушеку удалось в тысячу раз очистить возбудитель скрепи от других белков в исходной суспензии мозга погибшего животного. В таких препаратах возбудитель скрепи неожиданно оказался высокочувствителен к действию протеаы К и трипсина, но не инактивировался нуклеазами.
 Стенли Прузинер 28.05.1942
Стенли Прузинер 28.05.1942
Было показано, что биологическая активность возбудителя скрепи связана с белком. Тут в исследования прионных болезней вступает Стенли Прузинер — американский биолог, сын эмигрантов из России. Исходя из полученных данных, Прузинер предположил для возбудителя скрепи и других родственных инфекций термин «прион». Дальнейшее усовершенствование очистки прионов, включающее фракционирование в градиенте плотности сахарозы, позволило авторам выявить, что белок прионов представлен молекулами одного вида.
Очистка в зональном роторе позволила установить, что большая часть инфекционного материала состоит из приона с молекулярной массой 27 — 30×103. По своей химической природе прионы представляют собой сиалогликопротеид, в котором к боковым цепям аминокислот присоединены остатки сахаров.
 Модель молекулы нормального приона
Модель молекулы нормального приона
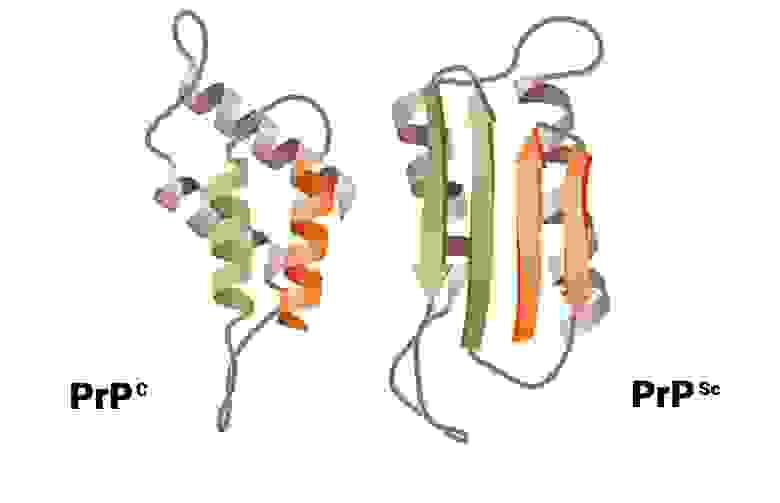
В ходе исследований было установлено, что возбудитель прионных заболеваний — мутированная (инфекционная) форма низкомолекулярного белка, названного прионным протеином (PrP). Прион (от англ. Proteinaceous infection particl) — белковоподобная инфекционная частица. Нормальная спиралевидная форма прионного протеина PrPС обнаружена в организме всех млекопитающих, в том числе и человека. Её кодирует единственный ген под названием PRNP, высокие уровни устойчивой экспрессии которого обнаруживают в нейронах в пятьдесят раз чаше, чем в глии.
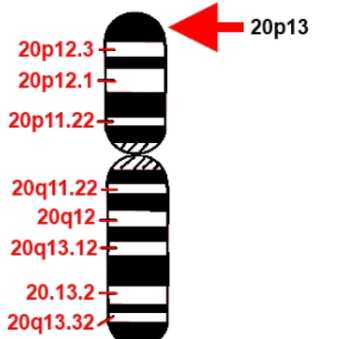 Ген PRNP локализован в коротком плече 20-й хромосомы
Ген PRNP локализован в коротком плече 20-й хромосомы
Ген PRNP, кодирующий PrP, обладает высокой степенью консервативности. Несмотря на это, в настоящее время установлены около сорока различных мутаций этого гена, которые связаны с различными прионными болезнями. В ряде случаев превращение нормального белка в прионный связано с мутациями этого гена. К таким заболеваниям относятся спорадичекая форма болезни Кройтцфельдта-Якоба, синдром Герстманна-Штреусслера-Шейнкера и фатальная семейная бессонница. Особую роль в этом имеют полиморфизм кодона 129, кодирующий метионин либо валин.
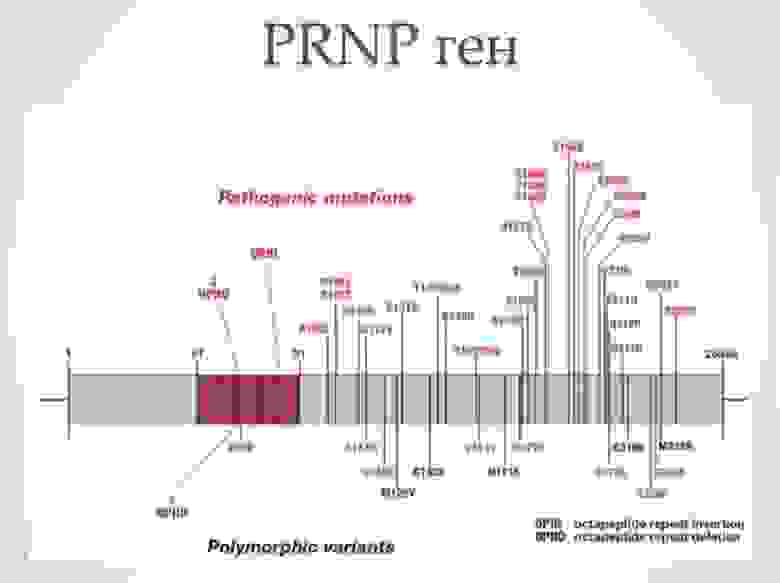 Известные мутации гена PNRP которые могут приводить к развитию прионных болезней.
Известные мутации гена PNRP которые могут приводить к развитию прионных болезней.
Среди заболевших болезнью Кройтцфельда-Якоба преобладают индивиды, гомозиготные по метионину либо валину, что позволяет предположить «защиту» от заболевания при гетерозиготности кодона 129. Было высказано предположение, что на агрегацию и конформационные изменения молекул PrP влияет аминокислота этого кодона. Эти данные получены на основании исследования членов племени форе.
В других случаях (новый вариант болезни Кройтцфельдта-Якоба и куру) инфекционный процесс развивается при заражении инфекционным прионом.
Нормальный белок PrPС участвует в передаче нервных импульсов в синаптических образованиях, играя определённую роль в регуляции суточных (циркадианных) циклов активности и покоя в клетках, органах и тканях, сохранении клеток Пуркинье, регуляции внутриклеточного содержания кальция в нейронах, поддержании трофики некоторых популяций и сохранением резистентности нейронов и астроцитов к повреждающим факторам. |
В 1991 году Прузинер предложил концепцию патогенеза губкообразных энцефалопатий, исходя из факта, что прионами человек инфицируется двумя путями:
Наследственной передачей по Менделю (аутосомно-доминантный тип наследования);
Не классическим, а последовательным наследованием, через предварительную генную ауторепликацию инфекционного агента.
Развитие болезни может происходить как за счёт точечных мутаций, так и в результате рекомбинантных событий. Мутации гена, кодирующего PrPС, программирует выработку другой, мутированной плоской формы — PrPSc с изменёнными аминокислотами. Таким образом, образование инфекционных прионовых белков происходит не за счёт репродукции молекулы PrPSc, попавшей в организм, а за счёт синтеза новых молекул, кодируемых мутировавшим геном PrPС.
Позже появились другие теории. Так, согласно одной из них, причина инфекционности PrPSc в следующем: инфекционный прион является зародышем для цепочечной полимеризации нормальных прионов. Конверсия PrPС в PrPSc представляет собой посттрансляционный процесс, включающий глубокое конформационное изменение, являющееся фундаментальным событием, лежащим в основе размножения инфекционных прионов. Эта форма обнаружена в организме людей и животных, больных прионными заболеваниями — трансмиссивными спонгиоформными (губкообразными) энцефалопатиями. В 1997 году за это открытие С. Прузинеру была вручена Нобелевская премия. Таким образом, установлено, что инфекционным агентом болезни Кройтцфельдта-Якоба, куру и скрепи служит белок.
Но как происходит, что вещество, относящееся к группе, поступающей в организм человека десятками и сотнями граммов за сутки, вызывает в организме такие разрушения?
Патогенез поражения обусловлен мутацией гена, кодирующего PrPС, программируют выработку другой, мутированной, плоской формы — PrPSc с изменёнными аминокислотами. В другом случае конверсия PrPС в PrPSc представляет собой посттрансляционный процесс, включающий глубокое конформационное изменение, являющееся фундаментальным событием, лежащим в основе размножения инфекционных прионов. Молекула PrPSc соединяется с молекулой PrPС с образованием димерного продукта, трансформирующегося с двумя молекулами PrPSc. В следующем цикле две молекулы PrPС соединяются с двумя молекулами PrPSc, что обеспечивает экспоненциальное образование молекул PrPSc. Белок PrPС — короткоживущий (период полураспада 5 — 6 часов), растворимый в жидких средах организма.
 Схема образования прионного белка и нерастворимых агрегатов (оригенал взят из книги И.Ю. Сергеев, В.А. Дубынин, А.А. Каменский «Физиология человека и животных. Том I)
Схема образования прионного белка и нерастворимых агрегатов (оригенал взят из книги И.Ю. Сергеев, В.А. Дубынин, А.А. Каменский «Физиология человека и животных. Том I)
В противоположность этому инфекционный белок PrPSc накапливается в цитоплазменных везикулах, что приводит к последующему нарушению функции синапсов и развитию глубоких неврологических дефектов. Позднее PrPSc высвобождается во внеклеточное пространство и откладывается в виде амилоидных бляшек. PrPSc форма обнаружена в организме людей и животных, больных прионными заболеваниями — трансмиссивными спонгиоформными (губкообразными) энцефалопатиями.
Поскольку этиологическим фактором развития спонгиозных энцефалопатий служит белок, исследователи столкнулись с единообразием патологических изменений ЦНС, что, в сущности, и послужило поводом для объёдинения этих заболеваний в единую группу.
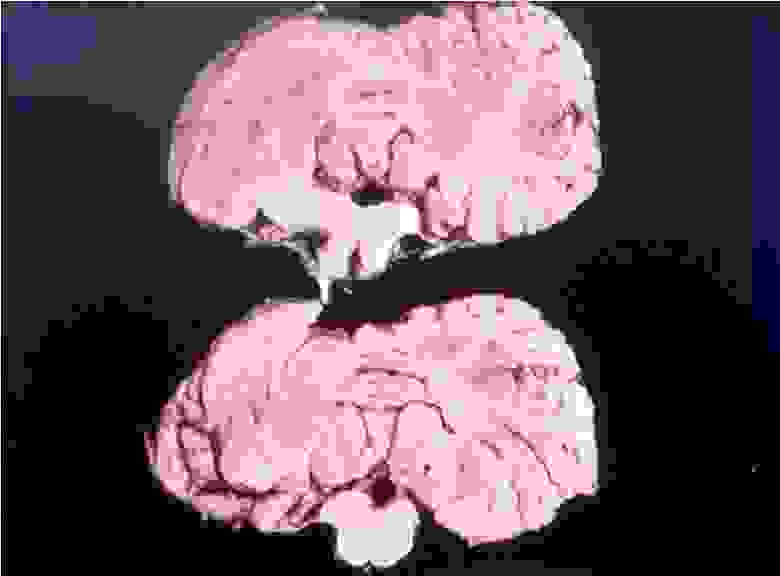 Мозг пациента погибшего от куру
Мозг пациента погибшего от куру
При болезни куру патологические изменения можно наблюдать только в центральной нервной системе. Обычно они выражаются в формировании типичной губчатой энцефалопатии. В коре, подкорковых ядрах, подбугорной области и в мозжечке наблюдается вакуолизация дендритов, аксонов и тел нейронов. Вакуолизация становится настолько выраженной, что серое вещество коры большого мозга приобретает вид губки, в результате чего такое состояние обозначают как status spongiosus. Методом электронной микроскопии было выяснено, что вакуоли окружены фрагментами таких же пролиферирующих мембран. Характерный патогистологический признак при куру — выпадение нейронов.
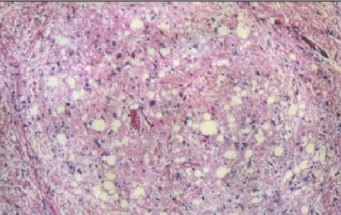 Гистологический срез
головного мозга
страдавшего от болезни
взрослого мужчины,
который впоследствии
погиб от куру
Гистологический срез
головного мозга
страдавшего от болезни
взрослого мужчины,
который впоследствии
погиб от куру
Изменения нейронов сочетаются с гипертрофией и размножением астроцитов. При куру у человека патогистологические изменения наиболее выражены в мозжечке: появляются аморфные ШИК-положительные бляшки, содержащие амилоид, уменьшается количество грушевидных нейронов (клеток Пуркинье). Вместе с тем, при куру не удаётся обнаружить воспалительной реакции, столь характерной для многих заболеваний ЦНС, или таких характерных признаков вирусной инфекции, как образование телец включений или глиальных узлов.
Характерные для куру изменения ЦНС обнаруживаются ещё до появления клинических симптомов заболевания, что сопровождается снижением общего содержания ганглиозидов в сером веществе головного мозга на 40%. Однако сам прион не удаётся обнаружить ни в крови, ни в сыворотке, ни в моче, ни в спинномозговой жидкости. В молоке, ткани плаценты и амниотической жидкости у людей или экспериментально заражённых животных прион также не обнаруживается.
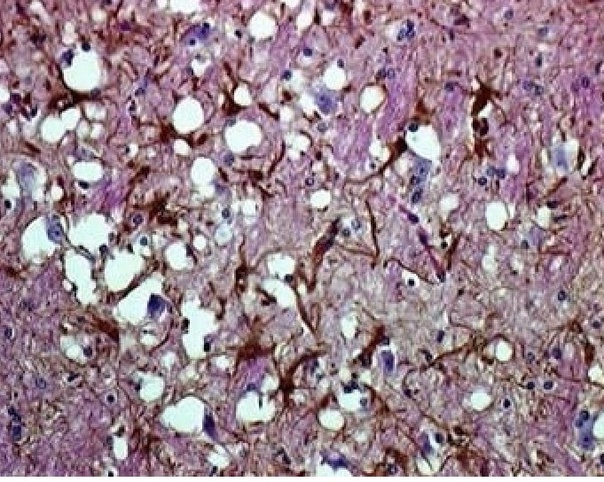 Гистологические измнения головного мозга овцы павшей от скрепи
Гистологические измнения головного мозга овцы павшей от скрепи
Таким образом, морфологически прионные болезни можно отнести к группе амилоидозов. Амилоидоз при прионных болезнях характеризуется отложением волокнистого амилоидного белка в головном мозгу. Образование прионных амилоидных бляшек сближает прионнные болезни с амилоидозами из группы нейродегенеративных заболеваний, таких, как, например, хорея Гентингтона. Получены убедительные доказательства того, что свойства прионных белков и амилоидов идентичны.
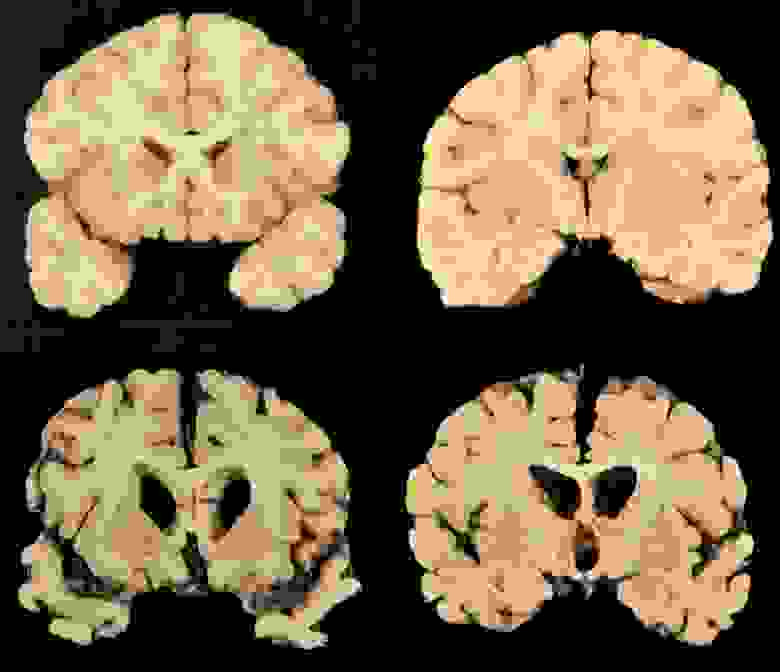 Нормальный мозг и мозг пациента с болезнью Кройтцфельда-Якоба (но дела у обоих пациентов, на самом деле, не очень)
Нормальный мозг и мозг пациента с болезнью Кройтцфельда-Якоба (но дела у обоих пациентов, на самом деле, не очень)
Тем не менее, при большом сходстве патогистологических изменений между болезнями Крейтцфельдта-Якоба, куру и скрепи имеются отличия. Так, амилоидные бляшки при болезни Кройцфельдта-Якоба наблюдаются в 9%, а при куру они встречаются в 70% случаев.
Уже давно было подмечено, что все симптомы при болезни Кройтцфельдта-Якоба подкрепляются патогистологическими изменениями. Так, при изменении моторных нейронов отмечено выпадение многих нервных клеток в двигательной области коры, включая гигантские пирамидные нейроны (клетки Беца), а также демиелинизацию пирамидных путей и утрату клеток передних рогов спинного мозга.
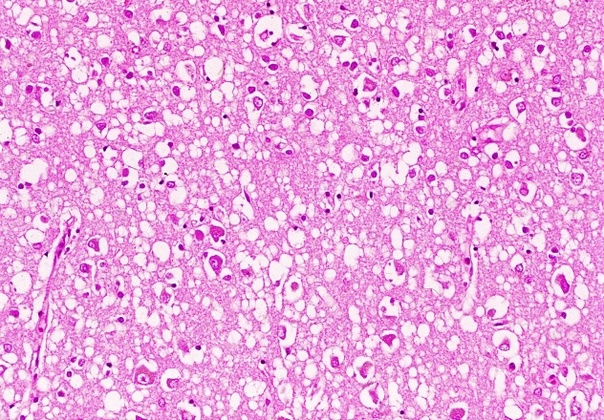 Гистологическая картина
болезни Кройтцфельда-Якоба
Гистологическая картина
болезни Кройтцфельда-Якоба
Уже с 70-х годов было установлено, что продолжительность инкубационного периода при куру в среднем составляет пять — десять лет. Однако при наблюдении среди лингвистической группы форе было установлено, что если заражение происходит в юности или раннем детском возрасте, то естественный инкубационный период при куру может продолжаться двадцать пять — тридцать лет. В организмах заражённых человека или обезьяны возбудитель куру накапливается в ткани головного мозга в наивысших концентрациях.
Эпидемиология прионных болезней весьма своеобразна. При куру источником инфекции является больной человек, точнее, ткани человека, погибшего от этого заболевания. Пути передачи возбудителя долгое время оставались неустановленными. Очень ценные данные были получены в экспериментах над обезьянами. Именно на обезьянах было выявлено, что куру развивается у этих животных не только в результате их внутримозгового заражения, но и после внутримышечного, подкожного или внутривенного введения заражённого материала. Важность этих наблюдений состоит в том, что они прямо свидетельствуют о необязательности попадания инфекционного агента непосредственно в мозг.

Гайдушек предположил, что передача приона связана с ритуалом людоедства, и поэтому специально изучил и подробно описал этот ритуал. Так, поедание умерших родственников, в котором главное участие принимали женщины и дети, рассматривалось как знак траура и уважения. Считалось, что с поеданием мозга умершего родственники приобретают его ум и все добродетели. Женщины и девушки голыми руками расчленяют человеческие трупы. Отделив мозг и мышцы, закладывают их голыми же руками в специально приготовленные бамбуковые цилиндры, которые затем недолго держат на раскалённых камнях в вырытых для этого в земле ямах.
 Аборигены форе несут больного куру ребёнка
Аборигены форе несут больного куру ребёнка
Гайдушек подчёркивал, что во время всей этой процедуры женщины обтирают руки о свои тела и волосы, чистят свои раны, расчёсывают места укусов насекомыми, вытирают детям глаза и носы. Проходит немного времени, и женщины и дети начинают толпиться вокруг очагов в нетерпеливом ожидании пиршества. Все дети, сами принимавшие участие в подобных пиршествах или имевшие матерей, участвовавших в таких ритуалах, обязательно оказываются заражёнными куру.
Главным аргументом в пользу доказательства алиментарного пути передачи инфекции служили результаты запрета с 1957 года ритуального людоедства среди народности форе, что привело к постепенному снижению заболеваемости в регионе. Так, в 1957 году было зарегистрировано двести двадцать случаев куру, а в 1966 — сто двадцать. |
В свою очередь, для того чтобы заболеть болезнью Кройтцфельдта-Якоба не обязательно съедать ближнего своего. Это заболевание, как правило, встречается в виде единичных (спорадических) случаев, однако примерно 10% представляют собой семейные очаги (каннибализм, однако, тут не при чём). Ранее существовало мнение, что семейные очаги являются результатом общности каких-то внешних воздействий, нежели наследственной передачи. Высказанное предположение подкреплялось примером одновременного заболевания тридцатидвухлетней женщины и её шестидесятипятилетнего мужа. Однако в следующее десятилетие семейные случаи болезни Кройтцфельдта-Якоба стали описываться чаще. Установлено, что они встречаются в 10 — 15% случаев этого заболевания. В этом отношении существенные сведения были представлены французскими исследователями, описавшими французскую семью, проживавшую в Арденах, в которой болезнь Кройтцфельдта-Якоба наблюдалась в трёх поколениях у восьми членов семьи. Авторы подчёркивали, что все члены этой семьи проживали в сельском доме в условиях тесного контакта друг с другом и их образ жизни характеризовался невысоким гигиеническим уровнем.
С самого начала исследований учёные обратили внимание на возможную роль плохо проваренного мяса, сырых морепродуктов, мозга свиней и коров в заражении людей болезнью Кройтцфельдта-Якоба. Оказалось, что заболевшие употребляли в пищу устриц и моллюсков значительно чаще по сравнению с контрольной группой. Исследователи высказали опасение, что употребление в пищу мозга свиней может таить в себе определённый риск заражения.
Тем не менее, кроме случаев заражения при употреблении в пищу мяса животных, были достоверно установлены случаи передачи вируса при операции по пересадке роговицы, и развитие у реципиента болезни Кройтцфельдта-Якоба через восемнадцать месяцев после операции. Описаны случаи заражения нейрохирурга и ещё трёх врачей, один из которых был стоматологом, а также двух пациентов этого зубного врача. Кроме того, в литературе можно встретить случай заражения двух молодых людей в результате вживления электродов, предварительно использованных для стереоэлектроэнцефалографии у пациента с верифицированной болезнью Кройтцфельдта-Якоба. Электроды оказались инфицированными, несмотря на предварительную стерилизацию в парах формалина. Это резко повысило интерес к распространению этого заболевания и особенно к возможности заражения при хирургических операциях и вскрытиях. К факторам риска относятся краниотомии, часто встречающиеся в анамнезе пациентов с болезнью Кройтцфельдта-Якоба.
Все случаи губчатоподобных энцефалопатий как у людей, так и у животных, в настоящее время заканчиваются фатально. Но недавно сотрудники Национального института аллергии и инфекционных заболеваний, и Института Броуда использовали антисмысловые олигонуклеотиды в борьбе с мышиной моделью скрепи. В этом случае мышей сначала заражали непосредственной инъекцией препарата гомогенезерованного мозга мышей, болеющих скрепи в терминальной стадии, а затем им вводили профилактическую инъекцию антисмысловых олигонуклеотидов к гену прионного белка PRNP непосредственно в желудочки мозга.
Предыдущие попытки использовать антисмысловые нуклеотиды проводились постоянным введением небольших доз и не принесли особого успеха. На этот раз исследователи применили два других метода. В первом случае мышам делалась однократная инъекция большой дозы препарата примерно на 120-й день жизни — незадолго до обычного времени первого появления у мышей симптомов признаков болезни. В этом случае развитие симптомов болезни удалось отодвинуть на 55% (в среднем на восемьдесят семь дней). Во второй серии опытов мышам делали инъекции каждые два — три месяца. Как отмечали авторы, в этом случае продолжительность жизни экспериментальных животных увеличивалась на 61 — 98%.
P.S. Если принять во внимание, что прионные болезни вообще неизлечимы, это очень неплохой результат. Так что надежда есть. До новых встреч! |

[1] Здесь и далее курсив мой. ˗˗ А.Л.
Список использованной литературы
Зуев В.А. Медленные вирусные инфекции человека и животных. — М.: Медицина,1988.
Зуев В.А. Завалишин И.А. Ройхель В.М. Прионные болезни человека и животных. — М.: Медицина, 1999.
Инфекционные болезни. Национальное руководство. /Под ред. Н.Д. Ющук Ю.Я. Венгеров. — М.: ГЭОТАР-Медиа, 2009.
Медицинская микробиология./ Под ред. В.И. Покровского. Изд. 3-е. — М.: Издательская группа ГЭОТАР-Медиа, 2005.
Основы патологии заболеваний по Роббинсону и Котрану. Т 3. — М.: Логосфера, 2016.
Покровский В.И. Пак С.Г. Брико Н.И. Данилкин Б.К. Инфекционные болезни и эпидемиология. — Изд. 2-е. — М: Издательская группа ГЭОТАР-Медиа , 2004.
Паевский А.С. Вот холера! — М.: АСТ, 2020.
Покровский В.И. Киселёв О.И. Черкасский Б.Л. Прионы и прионные болезни — М.: Изд-во РАМН, 2004.
Шувалова Е.П. Инфекционные болезни. — М.: Медицина, 2005.
