Задача N тел или как взорвать галактику не выходя из кухни
Не так давно я прочёл фантастический роман «Задача трёх тел» Лю Цысиня. В нём у одних инопланетян была проблема — они не умели, с достаточной для них точностью, вычислять траекторию своей родной планеты. В отличии от нас, они жили в системе из трёх звёзд, и от их взаимного расположения сильно зависила «погода» на планете — от испепеляющей жары до леденящего мороза. И я решил проверить, можем ли мы решать подобные задачи.
Физика явления
Для понимания задачи необходимо разобраться с физикой явления. В рамках классической теории сила притяжения двух тел определяется законом Ньютона:

где  — положение тел в пространстве,
— положение тел в пространстве,  — массы тел,
— массы тел,  — гравитационная постоянная.
— гравитационная постоянная.
В системе из  тел на каждое из них будет действовать сила притяжения от остальных, что выражается уравнением:
тел на каждое из них будет действовать сила притяжения от остальных, что выражается уравнением:

Воспользовавшись вторым законом Ньютона запишем ускорение для каждой частицы:

Вспоминая, что ускорение — это вторая производная координаты по времени, получим дифференциальное уравнение второго порядка в частных производных, которое необходимо решить для получения траектории каждого тела:

Здесь важно заметить, что сложность вычисления функции  равна
равна  и сильно возрастает с увеличением количества взаимодействующих тел.
и сильно возрастает с увеличением количества взаимодействующих тел.
Математика
Первым и простейшим методом решения дифференциальных уравнений является метод Эйлера, который предназначен для решения уравнений вида:

При переходе в дискретную область получим:

где  — шаг интегрирования, а
— шаг интегрирования, а  — число шагов интегрирования. Таким образом, ести нам необходимо произвести вычисление положения тел на момент времени
— число шагов интегрирования. Таким образом, ести нам необходимо произвести вычисление положения тел на момент времени  , то нам следует сделать
, то нам следует сделать  шагов интегрирования. Тут видна первая проблема — если велико, то нам нужно сделать большое количество шагов интегрирования.
шагов интегрирования. Тут видна первая проблема — если велико, то нам нужно сделать большое количество шагов интегрирования.
Для применения метода Эйлера к нашей задаче её следует свести к системе первого порядка. Для этого введём дополнительную переменную — скорость частицы:

Второй проблемой в решении систем дифференциальных уравнений является точность решения и её контроль. Точность можно повышать двумя способами: уменьшением шага интегрирования и выбором метода с более высоким порядком точности. Оба способа ведут к увеличению вычислительной сложности, но разными путями. Например, можно использовать классический метод Рунге-Кутты четвертого порядка, он требует четырёх вычислений функции на каждом шаге, но имеет порядок точности  (для сравнения, метод Эйлера имеет порядок точности
(для сравнения, метод Эйлера имеет порядок точности  и требует одного вычисления ). Контроль точности решения также можно осуществлять несколькими способами: сравнить с аналитическим решением, решить разними методами или с разным шагом и сравнить результаты, контролировать сторонние параметры и ограничения, которым должно соответствовать решение.
и требует одного вычисления ). Контроль точности решения также можно осуществлять несколькими способами: сравнить с аналитическим решением, решить разними методами или с разным шагом и сравнить результаты, контролировать сторонние параметры и ограничения, которым должно соответствовать решение.
Также, у каждого из этих методов есть свои недостатки. Аналитические решения могут отсутствовать, или, даже в большинстве случаев, и вовсе отсутствуют. Например, для нашей задачи тел аналитическое решение есть только при  , но даже этого достаточно для тестирования точности методов. Решение задачи двумя методами или с разным шагом увеличивает время вычислений, но этот подход возможно применять практически для любой задачи. Ограничения есть не у каждой задачи, но для нашей они есть: на каждом шаге интегрирования мы можем контролировать выполнение законов сохранения. Этот подход тоже увеличивает сложность вычисления, но здесь есть из чего выбирать, вычисление суммы импульсов или моментов импульса всех частиц имеет сложность порядка
, но даже этого достаточно для тестирования точности методов. Решение задачи двумя методами или с разным шагом увеличивает время вычислений, но этот подход возможно применять практически для любой задачи. Ограничения есть не у каждой задачи, но для нашей они есть: на каждом шаге интегрирования мы можем контролировать выполнение законов сохранения. Этот подход тоже увеличивает сложность вычисления, но здесь есть из чего выбирать, вычисление суммы импульсов или моментов импульса всех частиц имеет сложность порядка  , в то время как вычисление полной энергии системы имеет сложность порядка .
, в то время как вычисление полной энергии системы имеет сложность порядка .
слагаемых. Сложность в том, что все слагаемые имеют сильо разный порядок, и даже при вычислениях с двойной точностью не удаётся вычислить это значение с точностью, достаточной для сравнения на разных шагах. Для преодоления этой проблемы пришлось применить суммирование по алгоритму Кэхэна.
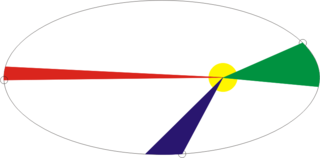
Рис 1. Пример эллиптической траектории.
Рассмотрим простой случай движения спутника по эллиптической орбите вокруг Земли. При приближении спутника к Земле его скорость увеличивается, а при удалении от Земли уменьшается, соответственно, напрашивается возможность уменьшать шаг интегрирования по времени на зелёном участке орбиты, и увеличивать на красном и синем без изменения точности решения. Попробуем сравнить более детально.
Таблица 1. Исследуемые методы решения дифференциальных уравнений
Для выбора наилучшего метода для нашей задачи произведём сравнение нескольких известных методов. Для этого смоделируем столкновение двух систем тел  и измерим относительное изменение полной энергии, импульса и его момента в конце моделирования (максимальное время моделирования
и измерим относительное изменение полной энергии, импульса и его момента в конце моделирования (максимальное время моделирования  ). При этом мы будем варьировать шаг и параметры методов интегрирования и замерять количество вызовов функции , соответственно, те методы, которые при меньшем числе вызовов приведут к меньшим потерям, будем считать более приемлимыми.
). При этом мы будем варьировать шаг и параметры методов интегрирования и замерять количество вызовов функции , соответственно, те методы, которые при меньшем числе вызовов приведут к меньшим потерям, будем считать более приемлимыми.
 |
 |
| a) | b) |
| Рис 2. Относительное изменение энергии a), импульса b), в конце моделирования системы тел различными методами в зависимости от количества вычислений функции с двойной точностью (double) |
Из графиков рисунке 2 видно, что наилучшее соотношение количества вычисления функции и относительного изменения энергии системы тел у методов Адамса пятого порядка и Дормана-Принса. Также видно, что для всех методов с ростом числа вычисления увеличивается относительное изменение импульса системы. Для относительного изменения энергии это также заметно, но только для нескольких методов, которые смогли достичь порога  . Этот эффект можно связать уже не с погрешностью методов, а с погрешностью вычислений, и дальнейшее увеличение точности возможно только совместно с увеличением точности вычислений с плавающей точкой.
. Этот эффект можно связать уже не с погрешностью методов, а с погрешностью вычислений, и дальнейшее увеличение точности возможно только совместно с увеличением точности вычислений с плавающей точкой.
 |
 |
| a) | b) |
| Рис 3. Относительное изменение энергии a), импульса b), в конце моделирования системы тел различными методами в зависимости от количества вычислений функции с четверной точностью (__float128) |
Из рисунков 3a и 3b видно, что применение вычислений с четверной точностью позволяет снизить относительные потери энергии вплоть до  , но нужно понимать, что время вычислений по сравнению с двойной точностью увеличивается на два порядка. Как и в случае с вычислениями с двойной точностью наилучшим соотношением точности и количества вычислений функции обладают методы Адамса пятого порядка и Дормана-Принса.
, но нужно понимать, что время вычислений по сравнению с двойной точностью увеличивается на два порядка. Как и в случае с вычислениями с двойной точностью наилучшим соотношением точности и количества вычислений функции обладают методы Адамса пятого порядка и Дормана-Принса.
Методы Дормана-Принса и Вернера относятся к классу вложенных методов и позволяют одновременно вычислить два решения с высоким и низким порядком точности (для метода Дормана-Принса порядки 5 и 4, а для метода Вернера порядки 6 и 5). Если эти два решения сильно отличаются, то мы можем разбить текущий шаг интегрирования на более мелкие. Что позволяет нам динамически изменять шаг интегрирования и уменьшать его только на тех участках, где это требуется.
Сравним методы Дормана-Принса, Вернера и Адамса пятого порядка более детально, на более длительном интервале моделирования нашей системы ( ).
).
 |
Рис 6. Относительное изменение энергии, импульса и его момента в процессе моделирования методом Адамса-Башфорта пятого порядка с шагом  |
 |
| Рис 7. Зависимости количества вычислений функции для методов Адамса пятого порядка, Дормана-Принса и Вернера от времени моделирования |
Видно, что на начальном этапе эволюции нашей системы ( ) все три метода показывают схожие характеристики, но на более поздних этапах в системе происходят некие события, в результате которых резко подскакивают ошибки в основных параметрах системы (полной энергии, импульса и его момента). Но методы Дормана-Принса и Вернера справляется с этими изменениями существенно лучше за счёт возможности уменьшать шаг интегрирования на «сложных» участках, в результате чего возрастает число вычислений функции , что видно на рисунках 4b и 5b, но общее число вычислений у вложенных методов меньше, чем у метода Адамса-Башфорта, что видно на рисунке 7.
) все три метода показывают схожие характеристики, но на более поздних этапах в системе происходят некие события, в результате которых резко подскакивают ошибки в основных параметрах системы (полной энергии, импульса и его момента). Но методы Дормана-Принса и Вернера справляется с этими изменениями существенно лучше за счёт возможности уменьшать шаг интегрирования на «сложных» участках, в результате чего возрастает число вычислений функции , что видно на рисунках 4b и 5b, но общее число вычислений у вложенных методов меньше, чем у метода Адамса-Башфорта, что видно на рисунке 7.
|
|
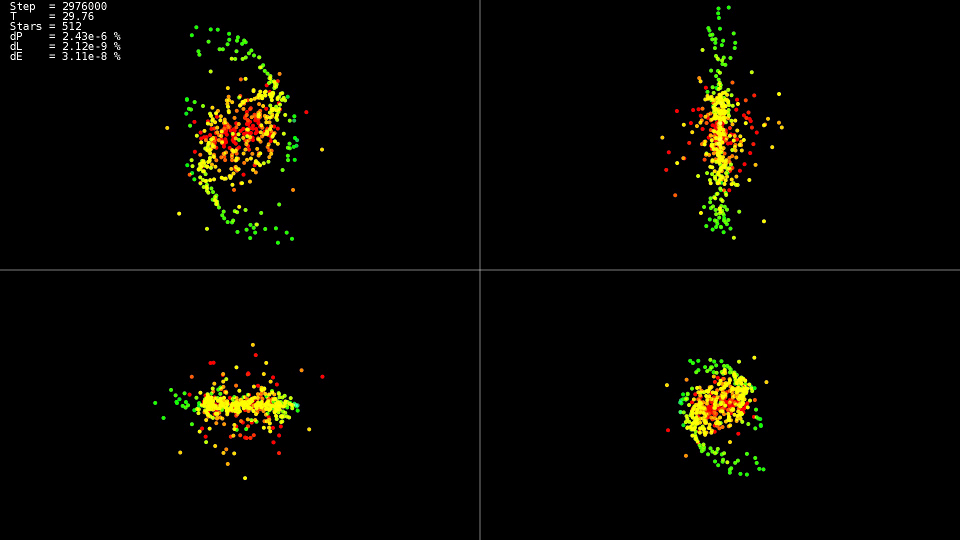
Видео 1. Моделирование системы из 512 тел. Метод Дормана-Принса. Динамический шаг  |
Видео демонстрирует, что до момента времени  движение относительно спокойное, а после происходит столкновение центров «галактик», которое приводит к резкому изменению траекторий и сильному увеличению скоростей некоторых частиц. При этом для сохранения точности решения необходимо уменьшать шаг интегрирования. Вложенные методы могут сделать это автоматически, на графиках видно, что на некоторых участках эволюции системы шаг интегрирования был уменьшнен почти на два порядка с
движение относительно спокойное, а после происходит столкновение центров «галактик», которое приводит к резкому изменению траекторий и сильному увеличению скоростей некоторых частиц. При этом для сохранения точности решения необходимо уменьшать шаг интегрирования. Вложенные методы могут сделать это автоматически, на графиках видно, что на некоторых участках эволюции системы шаг интегрирования был уменьшнен почти на два порядка с  до
до  . При использовании метода Адамса и такого шага на всём интервале эволюции системы мы не дождались бы решения.
. При использовании метода Адамса и такого шага на всём интервале эволюции системы мы не дождались бы решения.
Итог
Для решения лучше использовать вложеные методы, которые позволяют динамически контролировать шаг интегрирования, и уменьшать его только на «сложных» участках траектории. Не стоит гнаться за методами самого высокого порядка. Даже при использовании типа данных 'double' они не достигают своих потенциальных возможностей, использование же типов данных с большей точностью сильно увеличивает время решения задачи.
CPU реализация
Теперь, когда выбор метода решения уравнений определён, попробуем разобраться с вычислением силы взаимодействия для каждой частицы. Получим двойной цикл по всем частицам:
for(size_t body1 = 0; body1 < count; ++body1)
{
const nbvertex_t v1(rx[ body1 ], ry[ body1 ], rz[ body1 ]);
nbvertex_t total_force;
for(size_t body2 = 0; body2 != count; ++body2)
{
if(body1 == body2)
{
continue;
}
const nbvertex_t v2(rx[ body2 ], ry[ body2 ], rz[ body2 ]);
const nbvertex_t force(m_data->force(v1, v2, mass[body1], mass[body2]));
total_force += force;
}
frx[body1] = vx[body1];
fry[body1] = vy[body1];
frz[body1] = vz[body1];
fvx[body1] = total_force.x / mass[body1];
fvy[body1] = total_force.y / mass[body1];
fvz[body1] = total_force.z / mass[body1];
}
Силы притяжения для каждого тела вычисляются независимо, и, для задействования всех ядер процессора, достаточно перед первым циклом написать директиву OpenMP:
#pragma omp parallel for
for(size_t body1 = 0; body1 < count; ++body1)
Т.к. каждое тело взаимодействует с каждым, то для уменьшения количества взаимодействий процессора с ОЗУ и улучшения использования кэша у нас есть возможность загружать в кэш часть данных и использовать их многократно:
#pragma omp parallel for
for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE)
{
nbcoord_t x1[BLOCK_SIZE];
nbcoord_t y1[BLOCK_SIZE];
nbcoord_t z1[BLOCK_SIZE];
nbcoord_t m1[BLOCK_SIZE];
nbvertex_t total_force[BLOCK_SIZE];
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
size_t local_n1 = b1 + n1;
x1[b1] = rx[local_n1];
y1[b1] = ry[local_n1];
z1[b1] = rz[local_n1];
m1[b1] = mass[local_n1];
}
for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE)
{
nbcoord_t x2[BLOCK_SIZE];
nbcoord_t y2[BLOCK_SIZE];
nbcoord_t z2[BLOCK_SIZE];
nbcoord_t m2[BLOCK_SIZE];
for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2)
{
size_t local_n2 = b2 + n2;
x2[b2] = rx[local_n2];
y2[b2] = ry[local_n2];
z2[b2] = rz[local_n2];
m2[b2] = mass[n2 + b2];
}
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
const nbvertex_t v1(x1[ b1 ], y1[ b1 ], z1[ b1 ]);
for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2)
{
const nbvertex_t v2(x2[ b2 ], y2[ b2 ], z2[ b2 ]);
const nbvertex_t force(m_data->force(v1, v2, m1[b1], m2[b2]));
total_force[b1] += force;
}
}
}
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
size_t local_n1 = b1 + n1;
frx[local_n1] = vx[local_n1];
fry[local_n1] = vy[local_n1];
frz[local_n1] = vz[local_n1];
fvx[local_n1] = total_force[b1].x / m1[b1];
fvy[local_n1] = total_force[b1].y / m1[b1];
fvz[local_n1] = total_force[b1].z / m1[b1];
}
}
Дальнейшая оптимизация заключается в вынесении содержимого функции вычисления силы в основной цикл и исключении деления и умножения на массу тела m1[b1]. Кроме того, что мы немного сократили вычисления, компилятор на таком развёрнутом цикле сможет применить векторные инструкции процессора SSE и AVX.
#pragma omp parallel for
for(size_t n1 = 0; n1 < count; n1 += BLOCK_SIZE)
{
nbcoord_t x1[BLOCK_SIZE];
nbcoord_t y1[BLOCK_SIZE];
nbcoord_t z1[BLOCK_SIZE];
nbcoord_t total_force_x[BLOCK_SIZE];
nbcoord_t total_force_y[BLOCK_SIZE];
nbcoord_t total_force_z[BLOCK_SIZE];
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
size_t local_n1 = b1 + n1;
x1[b1] = rx[local_n1];
y1[b1] = ry[local_n1];
z1[b1] = rz[local_n1];
total_force_x[b1] = 0;
total_force_y[b1] = 0;
total_force_z[b1] = 0;
}
for(size_t n2 = 0; n2 < count; n2 += BLOCK_SIZE)
{
nbcoord_t x2[BLOCK_SIZE];
nbcoord_t y2[BLOCK_SIZE];
nbcoord_t z2[BLOCK_SIZE];
nbcoord_t m2[BLOCK_SIZE];
for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2)
{
size_t local_n2 = b2 + n2;
x2[b2] = rx[local_n2];
y2[b2] = ry[local_n2];
z2[b2] = rz[local_n2];
m2[b2] = mass[n2 + b2];
}
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
for(size_t b2 = 0; b2 != BLOCK_SIZE; ++b2)
{
nbcoord_t dx = x1[b1] - x2[b2];
nbcoord_t dy = y1[b1] - y2[b2];
nbcoord_t dz = z1[b1] - z2[b2];
nbcoord_t r2(dx * dx + dy * dy + dz * dz);
if(r2 < NBODY_MIN_R)
{
r2 = NBODY_MIN_R;
}
nbcoord_t r = sqrt(r2);
nbcoord_t coeff = (m2[b2]) / (r * r2);
dx *= coeff;
dy *= coeff;
dz *= coeff;
total_force_x[b1] -= dx;
total_force_y[b1] -= dy;
total_force_z[b1] -= dz;
}
}
}
for(size_t b1 = 0; b1 != BLOCK_SIZE; ++b1)
{
size_t local_n1 = b1 + n1;
frx[local_n1] = vx[local_n1];
fry[local_n1] = vy[local_n1];
frz[local_n1] = vz[local_n1];
fvx[local_n1] = total_force_x[b1];
fvy[local_n1] = total_force_y[b1];
fvz[local_n1] = total_force_z[b1];
}
}
Таблица 2. Зависимость времени вычисления (в секундах) функции от количества взаимодействующих тел для различных CPU реализаций
|
2048 | 4096 | 8192 | 16384 | 32768 |
|---|---|---|---|---|---|
| simple | 0.0425 | 0.1651 | 0.6594 | 2.65 | 10.52 |
| openmp | 0.0078 | 0.0260 | 0.1079 | 0.417 | 1.655 |
| openmp+block+optimization | 0.0037 | 0.0128 | 0.0495 | 0.194 | 0.774 |
- cистема: Debian 9, Intel Core i7–5820K (6 core)
- компилятор: gcc 6.3.0
Хорошо видно, что версия с поддержкой OpenMP ускоряется в шесть раз, ровно по количеству ядер, а оптимизированная версия быстрее ещё немногим более чем в два раза. Так что, при оптимизации не стоит рассчитывать только на параллелизм. Интересно, что при вычислениях в один поток (simple) процессор работал на частоте 3.6 ГГц, в параллельной версии (openmp) сбросил частоту до 3.4 ГГц, а в параллельной и оптимизированной (openmp+block+optimization) сбросил ещё до 3.3 ГГц, но это не помешало ей работать в 13.6 раз быстрее. Также видно, что рост времени вычисления с увеличением размера задачи квадратичен, и дальнейшее увеличение делает задачу нерешаемой за разумное время.
GPU реализация
Но возникает желание производить вычисления ещё быстрее. Есть несколько доступных направлений для ускорения: вычисление на GPU, аппроксимация функции . Сначала для произведения вычислений на GPU я выбрал технологию OpenCL. Для более удобной работы была использована библиотека CLHPP. Главным достоинством OpenCL является то, что код можно запускать и на процессоре, и на GPU, что упрощает написание и отладку, а также расширяет список железа для запуска. В отладке помогает инструмент Oclgrind, который в рантайме показывает неверные вызовы OpenCL API и проблемы при обращении к памяти.
OpenCL
Для начала работы с OpenCL необходимо получить список доступных платформ. Самые распространённые платформы представляют компании AMD, Intel и NVidia.
std::vector platforms;
cl::Platform::get(&platforms);
Далее, после выбора платформы, нужно выбрать вычислительное устройство, которое эта платформа представляет:
const cl::Platform& platform(platforms[platform_n]);
std::vector all_devices;
platform.getDevices(CL_DEVICE_TYPE_ALL, &all_devices);
И в заключение подготовительного этапа нужно создать контекст и очереди в рамках которых будет выделяться память и производиться вычисления. Например, контекст, объединяющий все вычислительные устройства выбранной платформы, создаётся следующим образом:
cl::Context context(all_devices);
std::vector queues;
for(cl::Device device: all_devices)
queues.push_back(cl::CommandQueue(context, device));
Для загрузки исходного кода на вычислительное устройство его необходимо скомпилировать, для этого предназначен класс cl: Program.
std::vector< std::string > source_data;
cl::Program::Sources sources;
for(int i = 0; i != files.size(); ++i)
{
source_data.push_back(load_program(files[i]));//Загружаем из файла
sources.push_back(std::make_pair(source_data.back().data(),
source_data.back().size()));
}
cl::Program prog(context, sources);
devices.push_back(all_devices);
prog.build(devices, options);
Для описания параметров функции (ядра), которая выполняется на вычислительном устройстве есть удолный шаблон cl: make_kernel.
typedef cl::make_kernel< cl_int, cl_int, //Block offset
cl::Buffer, //mass
cl::Buffer, //y
cl::Buffer, //f
cl_int, cl_int, //yoff,foff
cl_int, cl_int //points_count,stride
> ComputeBlock;
Далее все просто: объявляем переменную с типом ядра, передаём в неё скомпилированную программу и имя вычислительного ядра, можем запускать ядро почти как обычную функцию.
ComputeBlock fcompute(prog, "ComputeBlockLocal");
cl::NDRange global_range(device_data_size);
cl::NDRange local_range(block_size);
cl::EnqueueArgs eargs(ctx.m_queue, global_range, local_range);
fcompute(eargs, ...все остальные аргументы); //Собственно, сам вызов ядра.
Само вычислительное ядро для OpenCL очень похоже на вариант 'openmp+block+optimization' для CPU, только в отличии от CPU версии управление первым циклом происходит при помощи OpenCL (диапазон цикла определяется переменной global_range из кода запуска ядра, а из ядра номер текущей итерации доступен с помощью функции get_global_id (0)). Сначала часть данных о телах загружается с локальную память, затем обрабатывается. Локальная память является общей для всех потоков в группе, поэтому загрузка происходит один раз для группы, а обрабатывается каждым потоком в группе и, т.к. локальная память существенно быстрее глобальной, вычисления происходят много быстрее.
__kernel void ComputeBlockLocal(int offset_n1, int offset_n2,
__global const nbcoord_t* mass,
__global const nbcoord_t* y,
__global nbcoord_t* f, int yoff,
int foff, int points_count, int stride)
{
int n1 = get_global_id(0) + offset_n1;
__global const nbcoord_t* rx = y + yoff;
__global const nbcoord_t* ry = rx + stride;
__global const nbcoord_t* rz = rx + 2 * stride;
__global const nbcoord_t* vx = rx + 3 * stride;
__global const nbcoord_t* vy = rx + 4 * stride;
__global const nbcoord_t* vz = rx + 5 * stride;
__global nbcoord_t* frx = f + foff;
__global nbcoord_t* fry = frx + stride;
__global nbcoord_t* frz = frx + 2 * stride;
__global nbcoord_t* fvx = frx + 3 * stride;
__global nbcoord_t* fvy = frx + 4 * stride;
__global nbcoord_t* fvz = frx + 5 * stride;
nbcoord_t x1 = rx[n1];
nbcoord_t y1 = ry[n1];
nbcoord_t z1 = rz[n1];
nbcoord_t res_x = 0.0;
nbcoord_t res_y = 0.0;
nbcoord_t res_z = 0.0;
__local nbcoord_t x2[NBODY_DATA_BLOCK_SIZE];
__local nbcoord_t y2[NBODY_DATA_BLOCK_SIZE];
__local nbcoord_t z2[NBODY_DATA_BLOCK_SIZE];
__local nbcoord_t m2[NBODY_DATA_BLOCK_SIZE];
// NB! get_local_size(0) == NBODY_DATA_BLOCK_SIZE
for(int b2 = 0; b2 < points_count; b2 += NBODY_DATA_BLOCK_SIZE)
{
int n2 = b2 + offset_n2 + get_local_id(0);
// Copy data block to local memory
x2[ get_local_id(0) ] = rx[n2];
y2[ get_local_id(0) ] = ry[n2];
z2[ get_local_id(0) ] = rz[n2];
m2[ get_local_id(0) ] = mass[n2];
// Synchronize local work-items copy operations
barrier(CLK_LOCAL_MEM_FENCE);
nbcoord_t local_res_x = 0.0;
nbcoord_t local_res_y = 0.0;
nbcoord_t local_res_z = 0.0;
for(int local_n2 = 0; local_n2 != NBODY_DATA_BLOCK_SIZE; ++local_n2)
{
nbcoord_t dx = x1 - x2[local_n2];
nbcoord_t dy = y1 - y2[local_n2];
nbcoord_t dz = z1 - z2[local_n2];
nbcoord_t r2 = (dx * dx + dy * dy + dz * dz);
if(r2 < NBODY_MIN_R)
{
r2 = NBODY_MIN_R;
}
nbcoord_t r = sqrt(r2);
nbcoord_t coeff = (m2[local_n2]) / (r * r2);
dx *= coeff;
dy *= coeff;
dz *= coeff;
local_res_x -= dx;
local_res_y -= dy;
local_res_z -= dz;
}
// Synchronize local work-items computations
barrier(CLK_LOCAL_MEM_FENCE);
res_x += local_res_x;
res_y += local_res_y;
res_z += local_res_z;
}
frx[n1] = vx[n1];
fry[n1] = vy[n1];
frz[n1] = vz[n1];
fvx[n1] = res_x;
fvy[n1] = res_y;
fvz[n1] = res_z;
}
CUDA
Реализация для платформы NVidia CUDA немного проще, чем OpenCL, нам не нужно самим создавать контекст устройства и управлять очередью выполнения (по крайней мере, пока мы не захотим сделать multi-GPU реализацию). Как и в случае с OpenCL, нам необходимо выделить память на GPU, скопировать в неё наши данные, и потом можно запускать вычислительное ядро. Детальнее о работе с CUDA можно прочитать здесь.
dim3 grid(count / block_size);
dim3 block(block_size);
size_t shared_size(4 * sizeof(nbcoord_t) * block_size);
kfcompute <<< grid, block, shared_size >>> (...параметры ядра...);
В отличие от OpenCL, в CUDA задаётся не полный диапазон итераций (в OpenCL реализации это global_range), а задаётся размер грида и размеры блока в гриде, соответственно немного меняется вычисление текущего номера тела, в остальном ядро очень похоже на OpenCL, за исключением других имён функций синхронизации и спецификатора для разделяемой памяти. Ещё полезной отличительной особенностью CUDA является то, что мы можем указывать необходимый размер разделяемой памяти при запуске ядра. Как и в OpenCL реализации, в начале каждого блока итераций мы копируем часть данных в разделяемую память и потом работаем с этой памятью из всех нитей блока.
__global__ void kfcompute(int offset_n2, const nbcoord_t* y, int yoff, nbcoord_t* f, int foff,
const nbcoord_t* mass, int points_count, int stride)
{
int n1 = blockDim.x * blockIdx.x + threadIdx.x;
const nbcoord_t* rx = y + yoff;
const nbcoord_t* ry = rx + stride;
const nbcoord_t* rz = rx + 2 * stride;
nbcoord_t x1 = rx[n1];
nbcoord_t y1 = ry[n1];
nbcoord_t z1 = rz[n1];
nbcoord_t res_x = 0.0;
nbcoord_t res_y = 0.0;
nbcoord_t res_z = 0.0;
extern __shared__ nbcoord_t shared_xyzm_buf[];
nbcoord_t* x2 = shared_xyzm_buf;
nbcoord_t* y2 = x2 + blockDim.x;
nbcoord_t* z2 = y2 + blockDim.x;
nbcoord_t* m2 = z2 + blockDim.x;
for(int b2 = 0; b2 < points_count; b2 += blockDim.x)
{
int n2 = b2 + offset_n2 + threadIdx.x;
// Copy data block to local memory
x2[ threadIdx.x ] = rx[n2];
y2[ threadIdx.x ] = ry[n2];
z2[ threadIdx.x ] = rz[n2];
m2[ threadIdx.x ] = mass[n2];
// Synchronize local work-items copy operations
__syncthreads();
nbcoord_t local_res_x = 0.0;
nbcoord_t local_res_y = 0.0;
nbcoord_t local_res_z = 0.0;
for(int n2 = 0; n2 != blockDim.x; ++n2)
{
nbcoord_t dx = x1 - x2[n2];
nbcoord_t dy = y1 - y2[n2];
nbcoord_t dz = z1 - z2[n2];
nbcoord_t r2 = (dx * dx + dy * dy + dz * dz);
if(r2 < NBODY_MIN_R)
{
r2 = NBODY_MIN_R;
}
nbcoord_t r = sqrt(r2);
nbcoord_t coeff = (m2[n2]) / (r * r2);
dx *= coeff;
dy *= coeff;
dz *= coeff;
local_res_x -= dx;
local_res_y -= dy;
local_res_z -= dz;
}
// Synchronize local work-items computations
__syncthreads();
res_x += local_res_x;
res_y += local_res_y;
res_z += local_res_z;
}
n1 += foff;
f[n1 + 3 * stride] = res_x;
f[n1 + 4 * stride] = res_y;
f[n1 + 5 * stride] = res_z;
}
Таблица 3. Зависимость времени вычисления (в секундах) функции от количества взаимодействующих тел для различных GPU реализаций и лучшей CPU реализации
|
4096 | 8192 | 16384 | 32768 | 65536 | 131072 |
|---|---|---|---|---|---|---|
| openmp+block+optimization | 0.0128 | 0.0495 | 0.194 | 0.774 | --- | --- |
| OpenCL+половинка NVidia K80 | 0.004 | 0.008 | 0.026 | 0.134 | 0.322 | 1.18 |
| CUDA+половинка NVidia K80 | 0.004 | 0.008 | 0.0245 | 0.115 | 0.291 | 1.13 |
В целом результат неутешительный, всего лишь в 5–6 раз быстрее, чем CPU реализация. Даже если мы будем вести расчёты на всей K80, то получим ускорение до 12 раз, но т.к. сложность задачи квадратична, то мы за разумное время сможем обрабатывать не 32768 взаимодействующих тел, а 131072, что только в 4 раза больше.
Аппроксимация функции
Если присмотреться к функции, которой задаётся сила притяжения двух тел, то видно, что она квадратично убывает с расстоянием. Поэтому мы можем точно вычислять силу взаимодействия между близкими телами, и приближённо между отдалёнными. Одним из известных подходов
является алгоритм treecode, предложенный Д. Барнсом и П. Хатом. В моделируемом пространстве строится октодерево, содержащее в своих листьях координаты и массы моделируемых тел. В родительских узлах содержится центр масс, общая масса дочерних узлов и радиус сферы, описанной вокруг тел дочерних узлов. Корень дерева содержит центр масс всех тел, их общую массу и радиус сферы, описанной вокруг них. При расчёте силы взаимодействия сначала считается расстояние до корня дерева, если отношение расстояния до узла к его радиусу больше некоторой константы  , то корень считается одним телом с координатами, равными координатам центра масс входящих в него тел, и массой, равной сумме масс дочерних узлов, если отношение меньше или равно , то процедура рекурсивно повторяется для каждого дочернего узла. Если достигается лист дерева, то сила взаимодействия считается обычным способом. Таким образом, если одно тело сильно удалено от компактной группы других тел, эта группа представляется для него в виде одного тела, и сила взаимодействия рассчитывается не до каждого тела, а только до одного тела. Благодаря этому сложность алгоритма уменьшается с до
, то корень считается одним телом с координатами, равными координатам центра масс входящих в него тел, и массой, равной сумме масс дочерних узлов, если отношение меньше или равно , то процедура рекурсивно повторяется для каждого дочернего узла. Если достигается лист дерева, то сила взаимодействия считается обычным способом. Таким образом, если одно тело сильно удалено от компактной группы других тел, эта группа представляется для него в виде одного тела, и сила взаимодействия рассчитывается не до каждого тела, а только до одного тела. Благодаря этому сложность алгоритма уменьшается с до  ценой некоторой потери точности.
ценой некоторой потери точности.
В своём подходе я решил использовать не октодерево, а kd-дерево т.к. оно проще в использовании и имеет более низкие накладные расходы на хранение по сравнению с октодеревом.
Вернёмся обратно к реализации на CPU. Узел kd-дерева можно представить в виде класса, содержащего указатели на левого и правого потомка и информацию о координатах и массе:
class node
{
node* m_left; //!< Левый потомок
node* m_right; //!< Правый потомок
nbvertex_t m_mass_center; //!< Координаты центра масс узла
nbcoord_t m_mass; //!< Масса узла
nbcoord_t m_radius_sqr; //!< Квадрат радиуса описанной сферы, умноженный на lambda_crit
nbvertex_t m_bmin; //!< Минимальные координаты описанного бокса
nbvertex_t m_bmax; //!< Максимальные координаты описанного бокса
size_t m_body_n; //!< Номер тела, связанного с узлом
};
При таком способе хранения дерева у нас имеется два возможных варианта обхода дерева: либо использовать явную рекурсию, либо использовать стек самим. Я остановился на втором варианте.
nbvertex_t force_compute(const nbvertex_t& v1,
const nbcoord_t mass1)
{
nbvertex_t total_force;
node* stack_data[MAX_STACK_SIZE] = {};
node** stack = stack_data;
node** stack_head = stack;
*stack++ = m_root;
while(stack != stack_head)
{
node* curr = *--stack;
const nbcoord_t distance_sqr((v1 - curr->m_mass_center).norm());
if(distance_sqr > curr->m_radius_sqr)
{//Узел достаточно далеко, вычисляем силу и пропускаем его детей
total_force += force(v1, curr->m_mass_center, mass1, curr->m_mass);
}
else
{// Узел слишком близко, запоминаем детей для последующей обработки
if(curr->m_right != NULL)
{
*stack++ = curr->m_right;
}
if(curr->m_left != NULL)
{
*stack++ = curr->m_left;
}
}
}
return total_force;
}
Как и в cлучае «точной» CPU реализации, функция вычисления силы вызывается для каждого тела. Цикл по всем телам может быть легко распараллелен с помощью директив OpenMP.
Но соседние итерации цикла в этом случае будут обращаться к совершенно разным частям дерева, что не даёт эффективно использовать кэш процессора. Для преодоления этой проблемы обход всех тел нужно производить не по исходному порядку, а по порядку, по которому тела расположены в листьях kd-дерева, тогда соседние итерации будут происходить для тел, находящихся близко в пространстве, и обходить дерево по почти одинаковым путям.
template
void traverse(Visitor visit)
{
node* stack_data[MAX_STACK_SIZE] = {};
node** stack = stack_data;
node** stack_head = stack;
*stack++ = m_root;
while(stack != stack_head)
{
node* curr = *--stack;
if(curr->m_radius_sqr > 0)
{//Это не лист. Откладываем детей на стек.
if(curr->m_left != NULL)
{
*stack++ = curr->m_left;
}
if(curr->m_right != NULL)
{
*stack++ = curr->m_right;
}
}
else
{// Это листовой узел. Вычисляем силу.
visit(curr->m_body_n, curr->m_mass_center, curr->m_mass);
}
}
}
Эта реализация имеет другую проблему — нет универсального способа распараллелить такой обход дерева. Но мы можем полностью изменить способ хранения дерева в памяти — мы можем хранить все узлы в одном линейном масиве и полностью отказаться от хранения указателей на потомков, по аналогии с построением двоичной кучи. При начале нумерации узлов с  , если узел находится в ячейке номером
, если узел находится в ячейке номером  , то его левый потомок находится в ячейке
, то его левый потомок находится в ячейке  , правый потомок в ячейке
, правый потомок в ячейке  , а родитель в ячейке
, а родитель в ячейке  . Правый узел, соответствующий левому с номером , будет иметь номер
. Правый узел, соответствующий левому с номером , будет иметь номер  . Сам массив будет иметь длину
. Сам массив будет иметь длину  , а все листовые узлы будут расположены в последних ячейках, причём, близкие в пространстве узлы будут расположены в&n
, а все листовые узлы будут расположены в последних ячейках, причём, близкие в пространстве узлы будут расположены в&n
