Обзор архитектуры AlphaFold 2
В данном обзоре мы подробно рассмотрим нейронную сеть AlphaFold 2 от компании DeepMind, с помощью которой недавно был совершен прорыв в одной из важных задач биологии и медицины: определении трехмерной структуры белка по его аминокислотной последовательности.
В первых трех разделах обзора описывается задача, формат входных данных и общая архитектура AlphaFold 2. Далее, начиная с раздела «Input feature embeddings», описываются детали архитектуры. В разделе «Резюме» кратко суммируется основная информация из обзора.
В научной статье, опубликованной в Nature, и дополнительных материалах к ней, авторы используют название «AlphaFold» без цифры 2, и мы также будем его придерживаться.
Белки и их структуры
Белки — это органические молекулы, структура которых показана на рис. 1. Символом R обозначены аминокислотные остатки, которые могут быть 20 разных типов. Таким образом, белок можно закодировать строкой, записанной алфавитом из 20 символов.
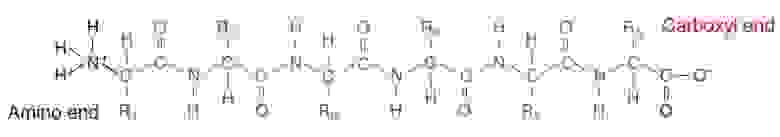 Рис. 1. Структура белка.
Рис. 1. Структура белка.
Белки сворачиваются в структуру за счет различных взаимодействий между атомами (водородные связи, ковалентные связи и др.). Структура белка определяется его аминокислотной последовательностью и, в свою очередь, определяет свойства этого белка в живом организме.
Задача, которую решает AlphaFold, заключается в предсказании структуры белка по его аминокислотной последовательности. Экспериментальное определение структуры белков является очень трудоемким. Решение этой задачи с помощью физического моделирования требует огромных вычислительных ресурсов. Проблема усугубляется тем, что в процессе сворачивания белка часто участвуют другие белки.
Каждые 2 года организуется соревнование CASP (Critical Assessment of protein Structure Prediction), в котором научные группы соревнуются в точности предсказания структур белков. Для оценки точности каждый раз используется новый набор белков, структуры которых уже были получены экспериментально, но еще не были опубликованы.
В 2020 году DeepMind с их нейронной сетью AlphaFold 2 выиграла соревнование CASP14, достигнув беспрецедентного уровня точности (рис. 2). DeepMind также выложили на YouTube видео об этом историческом успехе, которое рекомендую посмотреть. Об архитектуре AlphaFold версии 2 и пойдет речь в этом обзоре.
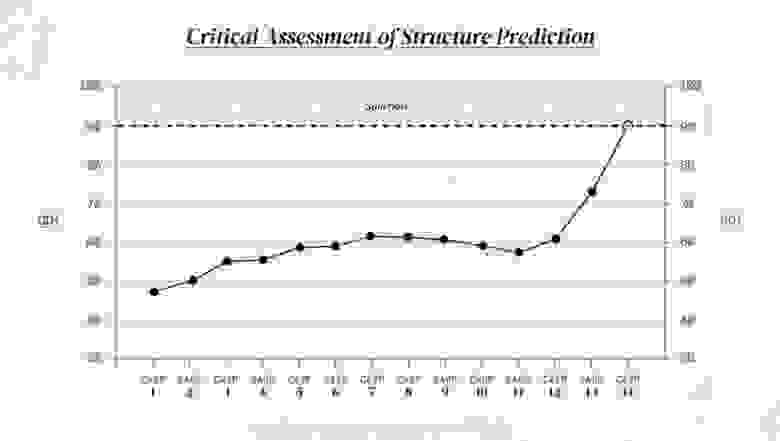 Рис. 2. Максимальная точность по метрике GDT, достигнутая в ходе соревнований CASP в разные годы (1994–2020).
Рис. 2. Максимальная точность по метрике GDT, достигнутая в ходе соревнований CASP в разные годы (1994–2020).
Предсказание структуры на основе эволюционного сходства
Аминокислотная последовательность белков меняется в процессе эволюции. Лишь часть аминокислот в цепочке влияет на структуру белка: мутации в этих местах скорее всего приведут к неправильному сворачиванию белка и утрате им своих полезных свойств. В результате организм с большой вероятностью отсеивается естественным отбором. Поэтому в ходе эволюции мутации в основном накапливаются в тех местах белка, которые не оказывают влияния на его структуру.
Если мы возьмем белок, выполняющий одну и ту же функцию в разных живых организмах, то увидим различия, накопившиеся в ходе эволюции от общего предка. Такое сопоставление называется множественным выравниванием последовательностей (multiple sequence alignment, MSA). Пример показан на рис. 3. Каждая строка — вид организма, столбец — код аминокислоты. Иногда в ходе эволюции аминокислоты могут удаляться или добавляться в белок. Чтобы было возможно выравнивание, удаленные аминокислоты в MSA обозначаются дефисом.
 Рис. 3. Пример таблицы MSA.
Рис. 3. Пример таблицы MSA.
Например, мы видим, что аминокислота на позиции 22 (лизин, символ «K») одинакова у всех организмов в таблице, тогда как аминокислота на позиции 23 сильно варьируется. Это говорит о том, что аминокислота К в данной позиции важна для сохранения структуры белка.
Мутации в местах, определяющих структуру белка, не только редки, но обычно происходят «парами»: то есть сразу два аминокислотных остатка, которые находятся в контакте друг с другом, мутируют так, что контакт сохраняется, а значит сохраняется структура белка и его свойства (рис. 4). В результате организм получается жизнеспособным и не отсеивается естественным отбором.
Рис. 4. Корреляции в таблице MSA.
Таблицу MSA можно составить для любого белка, найдя в базе данных наиболее схожие и эволюционно близкие к нему белки. Коррелирующие элементы последовательности вероятно контактируют, часто меняющиеся — не влияют на структуру. Поэтому таблицу MSA можно использовать при предсказании трехмерной структуры белка. Более того, для многих белков уже известны трехмерные структуры. Если в MSA найдутся белки с известными структурами, то по их структуре можно попытаться восстановить структуру исследуемого белка.
Предварительная обработка данных
Формат входных данных
Белок, для которого требуется определить структуру, будем называть целевым белком. Его аминокислотная последовательность представлена в виде строки из символов. Для целевого белка выполняется поиск в базе данных и составляется таблица MSA. Также происходит поиск шаблонов (templates): нескольких наиболее похожих белков с известной структурой. Каждый шаблон представлен в виде координат атомов в пространстве. Как вариант, может не быть ни одного шаблона. Таким образом, входными данными являются:
Целевой белок
Таблица MSA
Шаблоны (опционально)
Целевой белок
Целевой белок состоит из последовательности аминокислотных остатков (residues), для которых нужно определить пространственные положения.
Для целевого белка выполняется one-hot кодирование (20 аминокислот + «неизвестно»). Таким образом, аминокислотная последовательность длиной  превращается в массив
превращается в массив  из нулей и единиц размером
из нулей и единиц размером  .
.
Обрезка целевого белка и таблицы MSA
По оси, соответствующей номеру аминокислоты, целевой белок и таблица MSA обрезаются до фиксированной длины (выбирается случайный участок таблицы). На разных этапах обучения AlphaFold размер вырезаемого участка равен 256 и 384.
Создается массив  , который хранит для целевого белка позиции аминокислот до обрезки. Например, если мы обрезали белок с 20 по 40 позицию, то массив
, который хранит для целевого белка позиции аминокислот до обрезки. Например, если мы обрезали белок с 20 по 40 позицию, то массив  будет состоять из чисел [20, 21, …, 39].
будет состоять из чисел [20, 21, …, 39].
Подробнее см. Supplementary Material, раздел 1.2.8
Примечание. Здесь остается вопрос: насколько точно можно предсказать структуру белка, если обрезать его часть? В работе я не нашел освещения этого вопроса, но точность AlphaFold говорит сама за себя. Мне удалось найти таблицу распределения белков по длинам аминокислотных последовательностей (рис. 5).
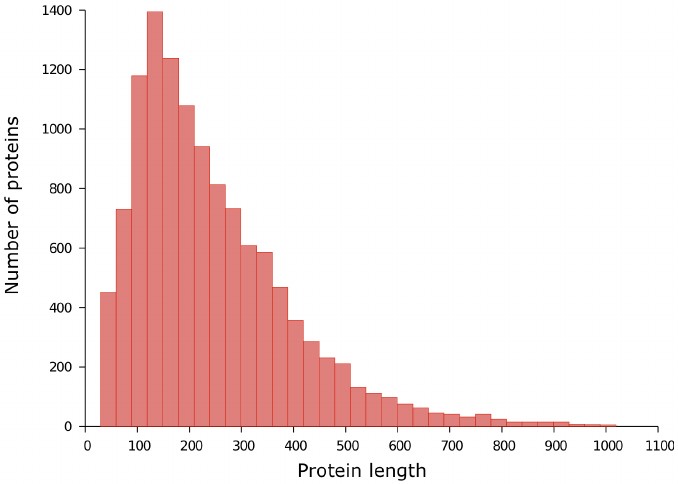 Рис. 5. Распределение белков по длинам аминокислотных последовательностей в базе данных PDB30.
Рис. 5. Распределение белков по длинам аминокислотных последовательностей в базе данных PDB30.
Кластеризация и маскирование таблицы MSA
Сложность вычислений и объем требуемой памяти в AlphaFold квадратично зависит от количества последовательностей в MSA, и лишь линейно от длины последовательности, поэтому количество последовательностей в MSA желательно уменьшить. Можно было бы выбирать случайную подвыборку последовательностей, но авторы предлагают другой подход: последовательности объединяются в кластеры.
В качестве метрики расстояния выбирается расстояние Хэмминга между последовательностями: число позиций, в которых аминокислоты различаются. Случайная подвыборка последовательностей выбирается в качестве центров кластеров, для всех остальных последовательностей ищется ближайший кластер.
Также 15% аминокислот в MSA «маскируется», то есть заменяется на случайную аминокислоту или на символ [MASK], для которого также выделяется бит в one-hot кодировании. Одна из задач сети AlphaFold 2 в ходе обучения состоит в том, чтобы предсказать замененные аминокислоты. Это работает аналогично задаче «masked language model» в языковой модели BERT.
Подробнее см. Supplementary Material, раздел 1.2.7
Если количество кластеров равно  , а длина последовательности равна
, а длина последовательности равна  , то данные собираются в массив
, то данные собираются в массив  размером (
размером ( ,
,  , 49). Для каждого
, 49). Для каждого  ,
,  вектор
вектор  размерностью 49 является конкатенацией one-hot кодирования
размерностью 49 является конкатенацией one-hot кодирования  -й аминокислоты центра
-й аминокислоты центра  -го кластера, распределения по аминокислотам для всего
-го кластера, распределения по аминокислотам для всего  -го кластера и некоторых дополнительных данных.
-го кластера и некоторых дополнительных данных.
Таким образом, массив  содержит информацию о таблице MSA, после кластеризации, маскирования и one-hot кодирования.
содержит информацию о таблице MSA, после кластеризации, маскирования и one-hot кодирования.
На этом, однако, сложности не заканчиваются. Создается еще один дополнительный массив  , состоящий из дополнительного набора последовательностей из MSA, не включенных в кластеры. Аминокислоты также кодируются one-hot кодированием, добавляются некоторые дополнительные данные. В результате массив
, состоящий из дополнительного набора последовательностей из MSA, не включенных в кластеры. Аминокислоты также кодируются one-hot кодированием, добавляются некоторые дополнительные данные. В результате массив  имеет размер (
имеет размер ( ,
,  , 25).
, 25).
Подробнее см. Supplementary Material, раздел 1.2.9
Данные о шаблонах
Исходными данными служат координаты атомов  -углерода (или
-углерода (или  -углерода для аминокислоты глицин) (рис. 6).
-углерода для аминокислоты глицин) (рис. 6).
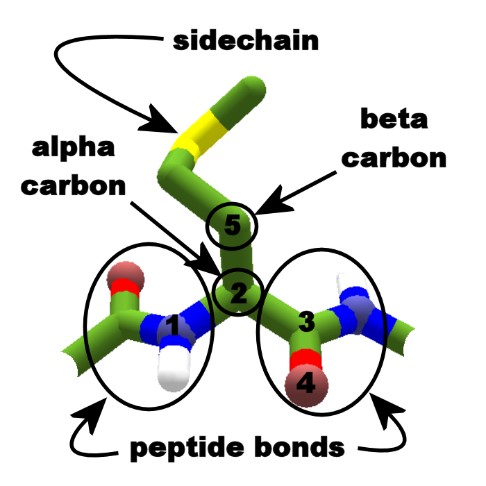 Рис. 6. Атомы alpha- и beta-углерода в молекуле белка.
Рис. 6. Атомы alpha- и beta-углерода в молекуле белка.
Создается массив попарных расстояний (авторы используют термин »дистограмма», англ. distogram) между атомами  -углерода. Этот массив инвариантен к сдвигу, повороту и отражению системы координат. Далее выполняется дискретизация каждого расстояния между 3.25 Å и 50.75 Å с 39 возможными значениями (последнее значение — »50.75 Å или больше»). После дискретизации выполняется one-hot кодирование.
-углерода. Этот массив инвариантен к сдвигу, повороту и отражению системы координат. Далее выполняется дискретизация каждого расстояния между 3.25 Å и 50.75 Å с 39 возможными значениями (последнее значение — »50.75 Å или больше»). После дискретизации выполняется one-hot кодирование.
Массив  состоит из полученной дистограммы и некоторых дополнительных данных. Этот массив имеет размер (
состоит из полученной дистограммы и некоторых дополнительных данных. Этот массив имеет размер ( ,
,  ,
,  , 88), где
, 88), где  — количество шаблонов.
— количество шаблонов.
Массив  содержит информацию об аминокислотах, из которых состоят шаблоны, и об углах, под которыми соединены атомы в цепочке.
содержит информацию об аминокислотах, из которых состоят шаблоны, и об углах, под которыми соединены атомы в цепочке.
Подробнее см. Supplementary Material, раздел 1.2.9
Результаты обработки
В результате предварительной обработки данных мы получили 6 массивов. Их размеры и описания суммаризированы ниже.
 — данные об аминокислотах целевого белка
— данные об аминокислотах целевого белка — данные о том, какую часть целевого белка мы обрезали
— данные о том, какую часть целевого белка мы обрезали — данные о кластеризованной таблице MSA
— данные о кластеризованной таблице MSA — данные о дополнительных последовательностях в MSA
— данные о дополнительных последовательностях в MSA — данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне
— данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне — данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне
— данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне
Архитектура AlphaFold
В этом разделе мы рассмотрим основные вычислительные блоки, из которых состоит архитектура AlphaFold версии 2. Рассмотрим назначение каждого блока и формат передаваемых между ними данных, но пока не будем детально рассматривать внутреннее устройство блоков.
Модель AlphaFold способна выдавать ответ (трехмерную структуру белка) за один end-to-end запуск. Но авторы обнаружили, что можно улучшить качество предсказаний, запуская AlphaFold многократно (recycling iterations): каждую следующую итерацию модель использует выходные данные, полученные на предыдущей итерации, и обновляет предсказания. Это усложняет архитектуру, поэтому сначала рассмотрим более простой вариант без использования recycling iterations (Рис. 7).
 Рис. 7. Упрощенный вариант AlphaFold без recycling iterations.
Рис. 7. Упрощенный вариант AlphaFold без recycling iterations.
Input feature embeddings
Первый блок AlphaFold принимает на вход 6 массивов, которые мы получили в результате предварительной обработки данных. Этот блок выполняет последовательность операций с обучаемыми весами, и его выходными данными являются три массива:
MSA representation, размером
 . Вектора-эмбеддинги для каждой пары «позиция + номер кластера». Под «позицией» имеем в виду номер аминокислотного остатка.
. Вектора-эмбеддинги для каждой пары «позиция + номер кластера». Под «позицией» имеем в виду номер аминокислотного остатка.Pair representation, размером
 . Вектора-эмбеддинги для каждой пары позиций.
. Вектора-эмбеддинги для каждой пары позиций.Extra MSA representation, размером
 . Вектора-эмбеддинги для каждой пары «позиция + номер последовательности».
. Вектора-эмбеддинги для каждой пары «позиция + номер последовательности».
Это работает так же, как в задачах NLP (эмбеддинги слов). Входные данные, изначально представленные в понятном человеку формате, мы переводим в некий «внутренний формат», понятный только нейронной сети. Например, в массиве pair representation каждой паре позиций соответствует вектор размерностью 128.
Элемент  массива pair representation является вектором-эмбеддингом пары позиций
массива pair representation является вектором-эмбеддингом пары позиций  , или, иначе говоря, эмбеддингом ориентированного ребра, связывающего позиции
, или, иначе говоря, эмбеддингом ориентированного ребра, связывающего позиции  и
и  (рис. 8). При этом считается, что между каждой парой позиций есть два ориентированных ребра (полносвязный граф).
(рис. 8). При этом считается, что между каждой парой позиций есть два ориентированных ребра (полносвязный граф).
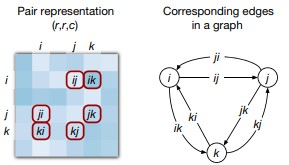 Рис. 8. Элемент pair representation как эмбеддинг направленного ребра.
Рис. 8. Элемент pair representation как эмбеддинг направленного ребра.
Evoformer stack
Evoformer-блок — это вычислительный блок с обучаемыми весами, разработанный для архитектуры AlphaFold. Этот блок принимает на вход массивы pair representation и MSA representation и возвращает два массива таких же размеров. Внутри Evoformer-блока происходит обмен информацией между массивами и их обновление.
В первых 4 evoformer-блоках осуществляется обмен информацией между pair representation и extra MSA representation. В следующих 48 блоках осуществляется обмен информацией между pair representation и MSA representation.
Внутреннее устройство evoformer-блока мы разберем позднее. Пока достаточно сказать, что Evoformer-блок близок к трансформеру (Vaswani et al. 2017) и использует механизм self-attention.
Именно с помощью evoformer-блоков AlphaFold определяет трехмерную структуру белка. Полученный на выходе обновленный массив pair representation теперь содержит информацию о трехмерной структуре, и задача следующего блока — извлечь эту информацию и построить саму структуру в явном виде.
Примечание. Для построения трехмерной структуры (с точностью до отражения) достаточно иметь матрицу попарных расстояний между позициями, то есть массив размером ( ,
,  ). Поскольку массив pair representation имеет в 128 раз больший размер, нейронной сети должно не составить труда закодировать в нем ту же информацию. Кроме того, в pair representation должна быть закодирована информация, позволяющая восстановить не только позиции атомов
). Поскольку массив pair representation имеет в 128 раз больший размер, нейронной сети должно не составить труда закодировать в нем ту же информацию. Кроме того, в pair representation должна быть закодирована информация, позволяющая восстановить не только позиции атомов  -углерода, но и позиции все остальных атомов углерода и азота в молекуле белка, а также оценить уверенность в предсказаниях.
-углерода, но и позиции все остальных атомов углерода и азота в молекуле белка, а также оценить уверенность в предсказаниях.
Еще одним выходом Evoformer stack является массив single representation: для этого каждый вектор первой строки массива MSA representation, полученного на выходе, обрабатывается полносвязным слоем. Смысл массива single representation в том, что он кодирует информацию о каждом остатке в отдельности, а не о каждой паре остатков.
На схеме AlphaFold, которая была приведена выше, авторы для краткости не показали первые 4 evoformer-блока (extra evoformer stack). Более подробная схема показана рис. 9.
 Рис. 9. Более детальная схема вычислений в упрощенном варианте AlphaFold без recycling iterations.
Рис. 9. Более детальная схема вычислений в упрощенном варианте AlphaFold без recycling iterations.
Structure module
После получения выходных данных evoformer stack работа с MSA-таблицей заканчивается. Блок structure module принимает на вход данные, полученные на выходе из evoformer stack:
single representation размером
 — вектора-эмбеддинги для каждой позиции
— вектора-эмбеддинги для каждой позицииpair representation размером
 — вектора-эмбеддинги для каждой пары позиций
— вектора-эмбеддинги для каждой пары позиций
Предполагается, что в этих массивах уже закодирована информация о возможной структуре белка. Остается построить саму структуру. В задаче предсказания структуры белка выходные данные могут иметь разный формат, например:
Вариант 1. Координаты всех атомов белка. Систему координат можно выбрать произвольно, поэтому такое представление не единственно. Из-за этого не так просто подобрать подходящую функцию потерь.
Вариант 2. Массив попарных расстояний между атомами. Такое представление инвариантно к сдвигу и вращению системы координат, однако оно также инвариантно к отражению системы координат. Но два зеркально отраженных белка — это не одно и то же.
Вариант 3. Углы между всеми атомами в цепочке. Такое представление тоже инвариантно к отражению системы координат. Кроме того, оно неустойчиво: вся структура сильно меняется при небольших изменениях углов.
В AlphaFold 1 нейросеть предсказывала попарные расстояния и углы между атомами в цепочке. Координаты атомов затем вычислялись на основе этих данных градиентным спуском.
В AlphaFold 2 нейросеть напрямую предсказывает координаты атомов, а также оценивает уверенность в предсказаниях. В качестве основной функции потерь используется Frame Aligned Point Error (FAPE). Эта функция инвариантна к смене системы координат как в предсказанных, так и в эталонных данных.
Более детально блок Structure module и функцию потерь FAPE мы будем рассматривать в соответствующем разделе.
Recycling
В общем виде механизм recycling может быть применен к моделям, которые способны уточнять приблизительный ответ, используя дополнительные данные, то есть к моделям следующего вида:

Инференс с recycling заключается в инициализации ответа нулями и запуске модели выбранное число ( ) раз, используя входные данные и предыдущий ответ.
) раз, используя входные данные и предыдущий ответ.

Recycling рассматривается лишь как дополнительный механизм, повышающий точность, поэтому мы хотели бы, чтобы даже после первой итерации модель выдавала бы ответ с хорошей точностью. В качестве функции потерь можно выбрать среднее значение ошибки по всем итерациям:

Если модель дифференцируема, то такую функцию потерь можно минимизировать напрямую. Однако это приведет к существенному перерасходу памяти и времени вычислений, по сравнению с однократным запуском модели. Авторы предлагают другой подход: на каждом шаге обучения выбирается случайное число  между 1 и
между 1 и  (общее для всех примеров в батче), модель запускается
(общее для всех примеров в батче), модель запускается  раз, и градиент ошибки распространяется только по последней итерации.
раз, и градиент ошибки распространяется только по последней итерации.

Подробнее см. Supplementary Material, раздел 1.10
Применение recycling в AlphaFold
AlphaFold использует в качестве входных данных шаблоны, то есть трехмерные структуры эволюционно схожих белков. Эти шаблоны играют роль гипотез о том, какой может быть структура белка. Выходными данными также является трехмерная структура, поэтому к AlphaFold может быть применен механизм recycling (рис. 10). Для этого блок input feature embeddings должен быть модифицирован таким образом, чтобы использовать не только 6 входных массивов данных, но и выходные и промежуточные данные, полученные на предыдущей итерации:
На первой итерации эти значения инициализируются нулями.
 Рис. 10. Общая схема AlphaFold с recycling iterations.
Рис. 10. Общая схема AlphaFold с recycling iterations.
Авторы экспериментально подтверждают, что добавление механизма recycling в AlphaFold улучшает точность предсказаний — особенно это проявляется на тех белках, для которых таблица MSA содержит недостаточно информации.
Еще одна (несущественная) деталь заключается в том, что во время инференса evoformer stack запускается параллельно 3 раза на разных подвыборках таблицы MSA, и полученные выходные данные усредняются.
См. также Supplementary Material, Algorithm 2
Теперь начнем подробное рассмотрение каждого блока архитектуры.
Input feature embeddings
Блок Input feature embeddings (рис. 11) принимает на вход следующий набор данных:
Массивы, полученные в результате предварительной обработки данных:
 — данные об аминокислотах целевого белка.
— данные об аминокислотах целевого белка. — данные о том, какую часть целевого белка мы обрезали.
— данные о том, какую часть целевого белка мы обрезали. — данные о кластеризованной таблице MSA.
— данные о кластеризованной таблице MSA. — данные о дополнительных последовательностях в MSA.
— данные о дополнительных последовательностях в MSA. — данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне.
— данные о шаблонах, в т. ч. попарные расстояния между атомами в каждом шаблоне. — данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне.
— данные о шаблонах: аминокислоты и углы между атомами в каждом шаблоне.
Данные с предыдущей итерации (recycling), которые на первой итерации заменяются массивами из нулей:
 — массив внутренних представлений (эмбеддингов) для каждой позиции.
— массив внутренних представлений (эмбеддингов) для каждой позиции. — массив внутренних представлений (эмбеддингов) для каждой пары позиций.
— массив внутренних представлений (эмбеддингов) для каждой пары позиций. — координаты атомов
— координаты атомов  -углерода (или
-углерода (или  -углерода для глицина) в предсказанной моделью трехмерной структуре (см. также раздел «данные о шаблонах»).
-углерода для глицина) в предсказанной моделью трехмерной структуре (см. также раздел «данные о шаблонах»).
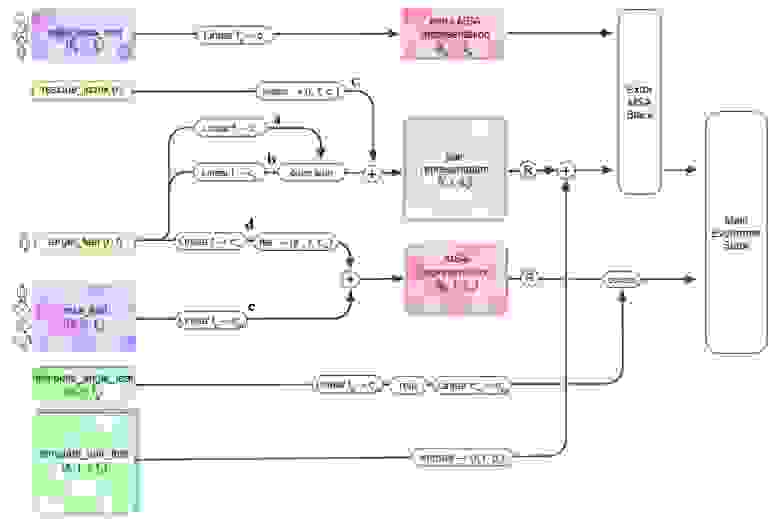 Рис. 11. Схема блока Input feature embeddings. Примечание: данные, полученные с предыдущей итерации, здесь не отображены; они используются в блоках «R» на данной схеме.
Рис. 11. Схема блока Input feature embeddings. Примечание: данные, полученные с предыдущей итерации, здесь не отображены; они используются в блоках «R» на данной схеме.
Общий смысл всех выполняемых действий в том, что информация, относящаяся по смыслу к одной позиции, идет в MSA representation, а информация, относящаяся по смыслу к паре позиций, идет в pair representation. Таким образом, в MSA representation собирается вся доступная информация о позициях, а в pair representation собирается вся доступная информация о парах позиций.
Pair representation
Начнем с того, как  и
и  преобразуются в
преобразуются в  .
.  — это последовательность векторов, соответствующих позициям в целевом белке. Каждый вектор состоит из нулей и только одной единицы (one-hot кодирование). Слой
— это последовательность векторов, соответствующих позициям в целевом белке. Каждый вектор состоит из нулей и только одной единицы (one-hot кодирование). Слой  переводит каждый вектор из исходной размерности в размерность
переводит каждый вектор из исходной размерности в размерность  . Применяя два таких слоя, мы получаем массивы
. Применяя два таких слоя, мы получаем массивы  и
и  .
.
Обработка вектора, полученного one-hot кодированием, с помощью полносвязного слоя — это то же самое, что слой Embedding в NLP-архитектурах. То есть каждой аминокислоте сопоставляется обучаемый вектор.
Чтобы получить pair representation, мы выполняем внешнюю сумму двух последовательностей векторов:

Далее с помощью массива  мы должны выполнить позиционное кодирование (как в трансформере). Для этого создается обучаемый вектор-эмбеддинг для чисел
мы должны выполнить позиционное кодирование (как в трансформере). Для этого создается обучаемый вектор-эмбеддинг для чисел  (всего 65 векторов). Вектор
(всего 65 векторов). Вектор  рассчитывается следующим образом: ищется число из множества
рассчитывается следующим образом: ищется число из множества  , ближайшее к
, ближайшее к  , и в качестве
, и в качестве  подставляется вектор-эмбеддинг, соответствующий этому числу.
подставляется вектор-эмбеддинг, соответствующий этому числу.

Таким образом, в позиционном кодировании стираются различия между всеми расстояниями меньше -32 и больше 32. Авторы комментируют это так:
Since we are clipping by the maximum value 32, any larger distances within the residue chain will not be distinguished by this feature. This inductive bias de-emphasizes primary sequence distances. Compared to the more traditional approach of encoding positions in the frequency space [Vaswani et al. 2017], this relative encoding scheme empirically allows the network to be evaluated without quality degradation on much longer sequences than it was trained on.
Полученные вектора позиционного кодирования прибавляются к  .
.
MSA representation
Для получения MSA representation мы используем  и набор MSA-кластеров
и набор MSA-кластеров  .
.
Аналогично, каждый вектор  и каждый вектор
и каждый вектор  обрабатываются полносвязными слоями, получая массивы
обрабатываются полносвязными слоями, получая массивы  и
и  . Однако массив
. Однако массив  получается двухмерный, а массив
получается двухмерный, а массив  трехмерный. Мы добавляем к массиву
трехмерный. Мы добавляем к массиву  новую ось, повторяя его столько раз, сколько было кластеров в MSA. Затем складываем полученные массивы.
новую ось, повторяя его столько раз, сколько было кластеров в MSA. Затем складываем полученные массивы.
Extra MSA representation мы получаем аналогичным способом, но уже без участия  .
.
Подробнее см. Supplementary Material, раздел 1.5
Использование информации о шаблонах
Каждый вектор в  , который содержит данные о шаблонах и углах в них, обрабатывается нейронной сетью со скрытым слоем (Linear-ReLU-Linear) для получения вектора-эмбеддинга, и полученный эмбеддинг конкатенируется с MSA representation.
, который содержит данные о шаблонах и углах в них, обрабатывается нейронной сетью со скрытым слоем (Linear-ReLU-Linear) для получения вектора-эмбеддинга, и полученный эмбеддинг конкатенируется с MSA representation.
Каждый вектор в  , обрабатывается полносвязным слоем для получения эмбеддинга (рис. 8, слой «embed»). Далее этот эмбеддинг обрабатывается цепочкой операций, которые не отражены на рис. 8, а именно:
, обрабатывается полносвязным слоем для получения эмбеддинга (рис. 8, слой «embed»). Далее этот эмбеддинг обрабатывается цепочкой операций, которые не отражены на рис. 8, а именно:
Triangular self-attention around starting node
Triangular self-attention around ending node
Triangular multiplicative update using outgoing edges
Triangular multiplicative update using incoming edges
Pair transition
Эти же операции применяются в evoformer-блоках, поэтому мы будем разбирать их позднее, при описании evoformer-блока. После применения этих операций мы получаем массив размером ( ,
,  ,
,  , 128) — эмбеддинг каждой пары позиций в каждом шаблоне.
, 128) — эмбеддинг каждой пары позиций в каждом шаблоне.
Затем мы добавляем эту информацию к pair representation, используя механизм template pointwise attention. Это вариант multi-head self-attention (Vaswani et al. 2017), в котором запросом (query) является  -й элемент pair representation, а ключами и значениями —
-й элемент pair representation, а ключами и значениями —  -е элементы каждого из шаблонов. Более детально останавливаться на этой операции сейчас не будем.
-е элементы каждого из шаблонов. Более детально останавливаться на этой операции сейчас не будем.
Подробнее см. Supplementary Material, раздел 1.7.1
Также в блоках «R» (recycling) мы используем информацию с предыдущей итерации. Для экономии места также не будем разбирать принцип работы блока «R», поскольку он является лишь дополнительным инструментом.
Подробнее см. Supplementary Material, раздел 1.10
Evoformer
Блок evoformer принимает и возвращает два массива:
MSA representation, размером
 — эмбеддинги пар (позиция + последовательность).
— эмбеддинги пар (позиция + последовательность).Pair representation, размером
 — эмбеддинги пар (позиция + позиция)
— эмбеддинги пар (позиция + позиция)
И устроен из блоков, имеющих собственные обучаемые веса (рис. 12).
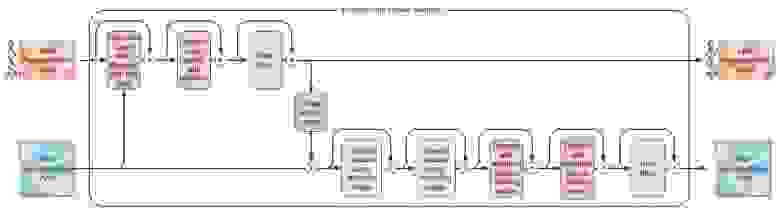 Рис. 12. Схема вычислений в evoformer-блоке.
Рис. 12. Схема вычислений в evoformer-блоке.
Evoformer похож на трансформер, поскольку многие операции в нем используют self-attention. Как в и трансформере, через все операции проброшены skip connections, то есть входные данные прибавляются к выходным. Операция, вокруг которой проброшен skip connection, называется residual-блоком. В некоторых residual-блоках добавляется dropout по отдельным столбцам и строкам. Общий алгоритм вычислений в evoformer stack, дублирующий рис. 12, приведен в алгоритме 6.

Большая часть операций, выполняемых в evoformer-блоке, основаны на criss-cross attention. Этот подход заключается в следующем: имея трехмерный массив, каждый  -й элемент которого является вектором-эмбеддингом, мы сначала применяем операцию multi-head self-attention к каждой строке массива, затем к каждому столбцу (или наоборот).
-й элемент которого является вектором-эмбеддингом, мы сначала применяем операцию multi-head self-attention к каждой строке массива, затем к каждому столбцу (или наоборот).
Надо так же отметить, что evoformer-блоки, обрабатывающие MSA-кластеры, и evoformer-блоки, обрабатывающие дополнительные MSA-последовательности (см. рис. 9, extra evoformer stack), имеют некоторые отличия. Эти отличия обусловлены тем, что дополнительные таблицы MSA содержат большое количество последовательностей (в отличие от MSA-кластеров), и обрабатывающие их evoformer-блоки должны быть адаптированы для работы с большим количеством последовательностей.
Подробнее см. Supplementary Material, раздел 1.7.2
Далее мы последовательно разберем все вычислительные элементы evoformer-блока (рис. 12).
MSA row-wise and column-wise gated self-attention
Вспомним, что массив MSA representation состоит из набора последовательностей, каждая из которых состоит из набора позиций. Каждая позиция п
