MiTCR – платформа для диагностики нового типа. Семинар в Яндексе
Рассказ Дмитрия Болотина посвящен программному обеспечению MiTCR, разработанному для анализа репертуаров иммунологических рецепторов. В своем докладе он рассмотрел основные особенности анализа сырых данных секвенирования, в частности, алгоритмы выравнивания последовательностей и коррекции ошибок в исходных данных, а также кратко описал архитектуру, производительность и ближайший план развития программы. Исходный код MiTCR открыт. В перспективе это ПО может вылиться в общую платформу для биоинформатиков, где они смогут обрабатывать свои данные и обмениваться ими с другими исследователями. Результатом такой совместной работы должен стать новый тип диагностики: при помощи анализа крови можно будет ответить не только на вопрос, есть ли у человека то или иное заболевание, а сразу определить, чем именно он болен.Видеозапись доклада
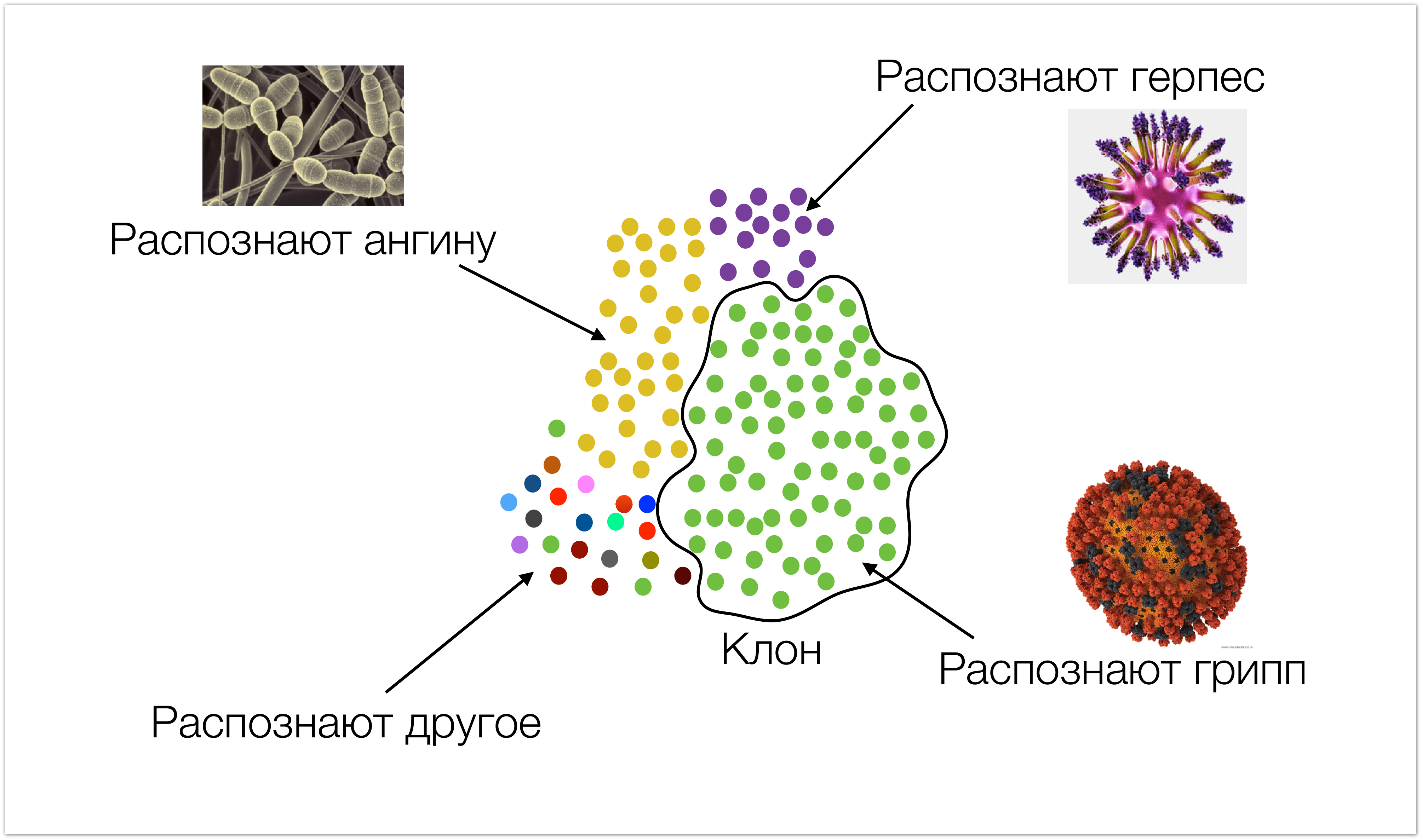
Начнем мы издалека, чтобы было понятно, с какими данными мы работаем, и откуда они берутся. На картинке ниже очень схематично изображен иммунитет. Одним цветом окрашены клетки, которые имеют одинаковую специфичность (т.е. они распознают одни и те же типы инфекций). Мы называем такие клетки клонами. Во время инфекционной атаки, количество клеток, которые ее распознают, возрастает.
Специфичность этих клеток обусловлена тем, что у них на поверхности есть Т-клеточный рецептор, правила сборки которого записаны в соответствующем гене. Для последующего повествования важно понимать его структуру.
Ген — это участок ДНК, можно о нем думать просто как о последовательности, состоящей из четырехбуквенного алфавита. Уникальность гена Т-клеточного рецептора заключается в том, что он разный в разных клетках. Все остальные гены, которые есть в наших клетках — одинаковые, отличаются только гены T-клеточного рецептора и антител, в T- и B-лимфоцитах соответственно.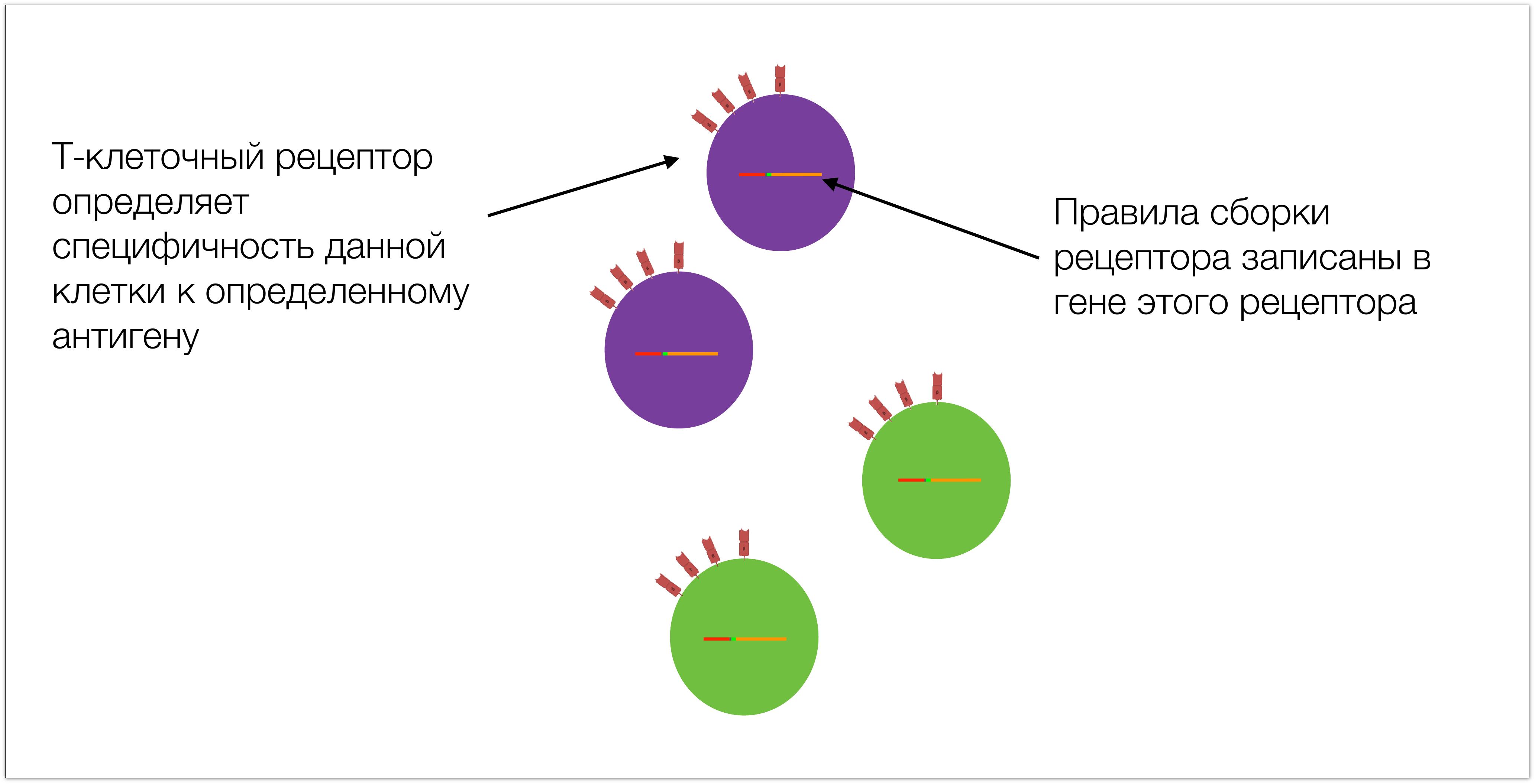
Состоит этот ген из 4х основных кусков. Один кусок в T-клеточных рецепторах всегда одинаковый. Два других участка (красный и зеленый на картинке) выбираются из небольшого набора. Во время сборки этих двух участков между ними добавляются случайные буквы (порядка десяти) что приводит к формированию гиганского разнообразия таких рецепторов. Тот участок, который потом в белке узнает антиген называется CDR3.
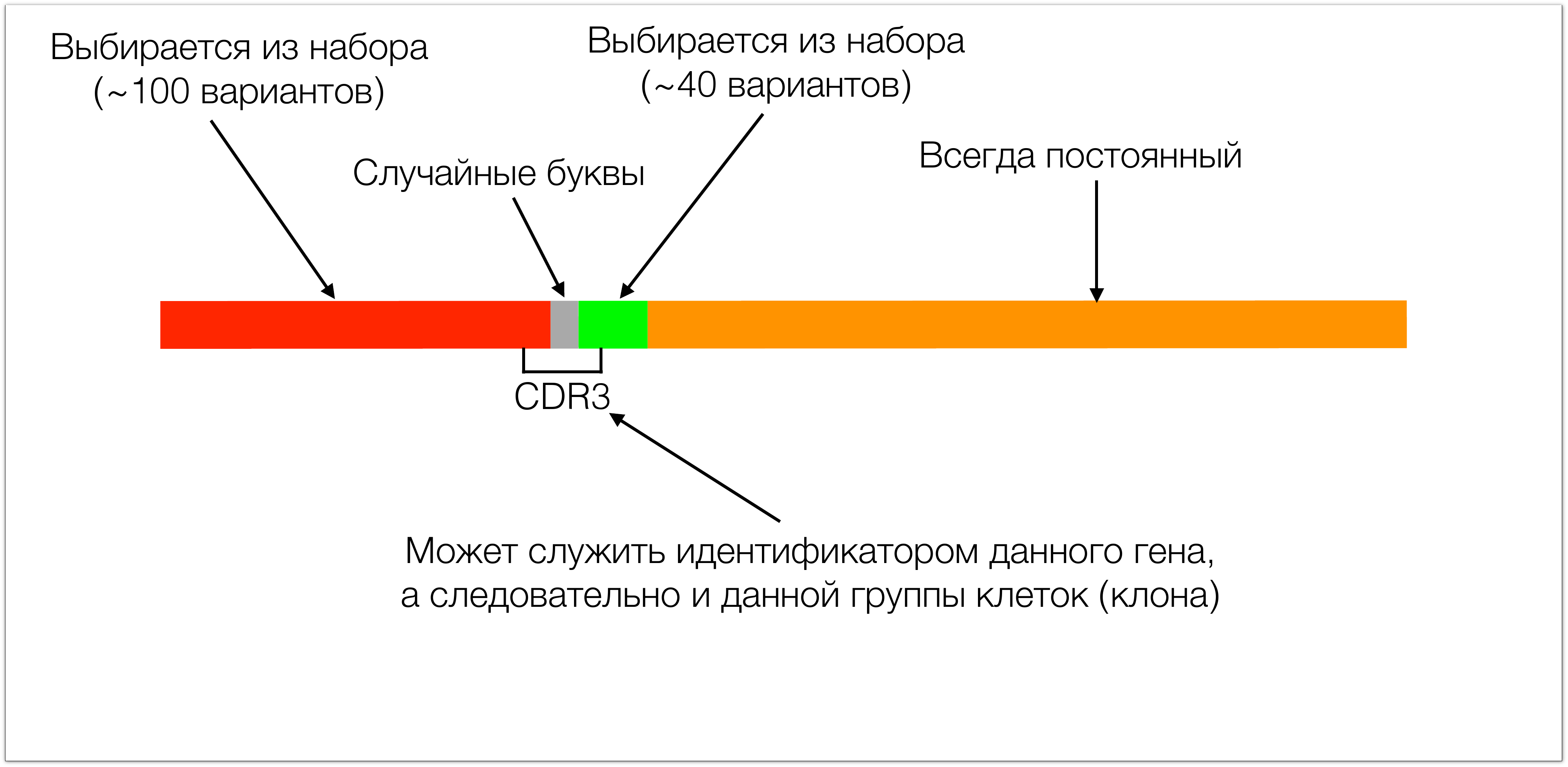
Он интересен с двух сторон: с одной стороны, он обуславливает специфичность, потому что просто по биологии он как раз кодирует ответственный за распознавание антигена участок рецептора, а с другой стороны он удобен как идентификатор, потому что в нем сосредоточено все разнообразие, в него входит кусок красного участка, кусок зеленого. По этим кускам мы полностью можем определить, какой из набора этих сегментов был выбран, и в нем полностью сосредоточены все эти случайные буквы. Поэтому, зная CDR3 мы можем реконструировать весь белок. Мы также можем его использовать как идентификатор, потому что, если CDR3 одинаковый, то и весь ген тоже одинаковый.
Итак, у нас есть образец крови, мы проводим некоторую последовательность сложных молеулярно-биологических реакций для того, чтобы выделить из него последовательности ДНК, содержащие CDR3. Далее мы загружаем их на секвенатор, а на выходе получаем что-то в этом роде:

Это реальные данные, которые к нам приходят из секвенсового центра. Если тут отбросить лишнее, то по сути это просто последовательности ДНК длинной от 100 до 250 нуклеотидов. И последовательностей таких очень много.
Наша задача реконструировать из этих данных то, что было в самом начале т.е. узнать, сколько и каких клонов было в первичном образце.
Все осложняется тем, что в данных есть самые разные ошибки. Ошибки эти вносятся на всех этапах: пока происходит пробоподготовка, пока секвенируется и т.д. Темпы появления ошибок могут быть разными, в зависимости от архитектуры эксперимента от секвенатора, который был использован, в общем, от всего. Это могут быть вставки, дилеции и замены.
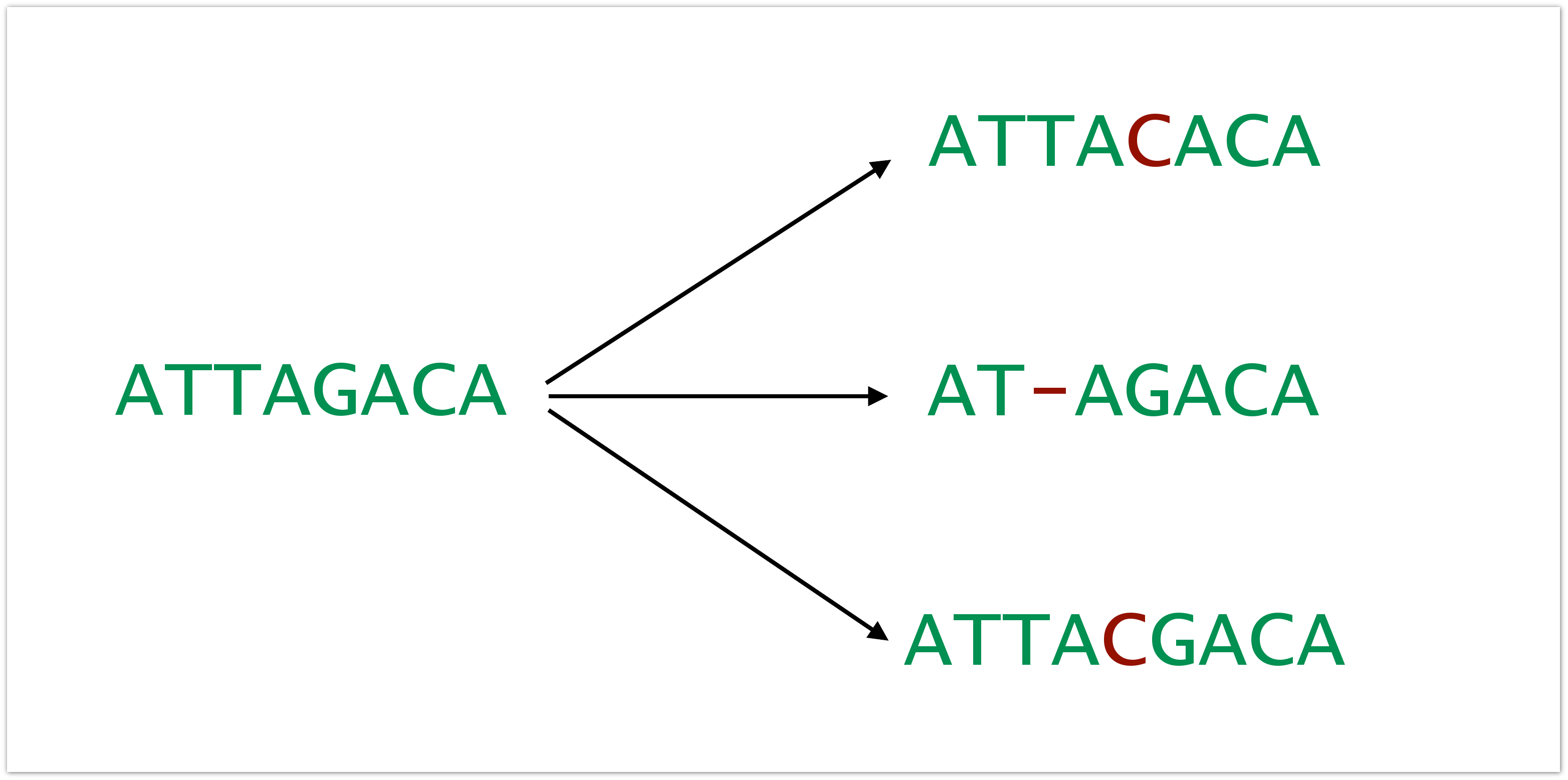
И это плохо, в первую очередь, с той точки зрения, что данные об образце искажаются и вносится искусственное разнообразие. Допустим у нас был образец, состоящий только из одного клона, но в результате подготовки в него внеслись ошибки. В результате мы видим, что у нас больше клонов, чем там было, и у нас складывается неверная картина о репертуаре Т-клеток у пациента.
Теперь коротко о том, с какими данными мы работаем. На входе мы получаем порядка ста миллионов сиквенсов, а то и миллиард, длиной по 100–250 нуклеотидов. Все это в сумме весит до 100 ГБайт. И концентрация CDR3 оно может быть от 1/5 (в совсем редких случаях бывает больше) и до считаных последовательностей миллионы.
Схема разработанного нами ПО MiCTR выглядит примерно следующим образом:

MiCTR решает проблему, которую я описал ранее: он из данных делает список клонов и корректирует ошибки. Первый блок извлекает из данных CDR3, следующие блоки работают только уже с CDR3 и объединяют их в клоны и корректируют ошибки.
На первом блоке я не буду сильно концентрироваться. Он занимается выравниваниями, а это огромная часть биоинформатики, которая потребовала бы отдельной лекции. Если коротко, то этот блок размечает входные последовательности, определяя где в них красный, а где зеленый участки. Набор, из которых они выбираются известен, он в базах данных и абсолютно одинаков для всех людей. Мы определяем, какой из этих красных участков нашелся и где. То же самое проделываем с зелеными участками. Построив эти выравнивания, мы уже знаем, где у нас находится CDR3, и можем выделять их из последовательностей и работать с ними.
Самая банальная вещь, которую нужно сделать с этими данными, это объединить их по одинаковости: одинаковые CDR3 нужно собрать и их сосчитать. Но т.к. в данных присутствуют ошибки, которые нужно корректировать, и для их коррекции мы впоследствии будем осуществлять нечетки поиск по множеству CDR3 (например поиск CDR3, отличающихся на одну букву), то для хранения здесь мы используем префиксное дерево (trie), очень удобное для подобных опрераций. Кроме того, при росте это дерево не перестраивается, поэтому очень удобно на нем имплементировать всякие конкурентные алгоритмы. Не нужно сильно заботиться о каких-то хитрых синхронизациях. Таким деревом очень просто пользоваться, но у него есть и недостатки. Оно тяжеловесно и весит достаточно много в оперативке. Однако в реальных условиях выясняется, что это не представляет особой проблемы (удается уложится в 1–2), поэтому это идеальный контейнер для таких данных.
Что мы делаем по отношению к ошибкам? На самом деле, собираем все эти клоны мы только из части сиквенсов. Дело в том, что секвенатор, кроме того, что дает буквы, которые он прочитал, каждой позиции ставит значение качества, которое определяет вероятность ошибки. Т.е. секвенатор прочитал букву, но он в ней может быть уверен или нет. И для каждой позиции устанавливается вероятность ошибки. И мы тут делаем достаточно простой трюк: мы просто проводим некоторый порог вероятности ошибки, скажем, в одну сотую. Все буквы, которые имеют большую вероятность ошибки, мы называем плохими. Соответственно все CDR3, содержащие такую букву, мы тоже считаем плохими. Все это хранится в контейнере, который не только коллекционирует данные по численности, но и собирает другую информацию: он ищет эквивалентные CDR3, либо CDR3 отличающиеся только по позициям с плохим качеством. Таким образом удается скорректировать большую долю ошибок. К сожалению ото всех ошибок так избавиться не получается, часть ошибок вносится еще до секвенатора, поэтому о них нет никакой дополнительной информации.
И тут мы делаем достаточно простую вещь, мы находим пары CDR3, которые отличаются на одну букву и сильно отличаются по концентрации. Чаще всего, это явный маркер ошибки. И мы просто удаляем такие их, выбирая порог порядка 1 к 10 по концентрации: одна ошибка на сиквенс.
В сумме все это позволяет нам скорректировать 95% ошибок и при этом не удалить естественное разнообразие. Понятно, что при таких алгоритмах можно ошибиться и принять естественный клон за ошибку, но по факту они так разнообразны, что этого не происходит. Такая производительность — это хорошие цифры для реальной работы.
Стоит немного описать программную реализацию всего этого. Система у нас много поточная, разметка последовательности запускается параллельно. В однопроцессорных системах наилучшего результата удается добиться, если количество потоков соответствует количеству ядер. На схеме выше отображена работа в два потока: один занимается формированием клонов из хороших CDR3, другой — из плохих. Все это неплохо масштабируется: например, на шестиядерном процессоре исполнение кода ускоряется примерно 3,5–4 раза. Что касается производительности, но на вполне обычном железе (AMD Phenom II X4 @ 3.2 GHz) обрабатывается 50 000 последовательностей в секунду. На хранение одного клонотипа уходит около 5 КБайт оперативки. Таким образом, самый сложный (разнообразный) массив из нашего опыта удалось обработать за 20 минут на машине с вышеозначенным процессором и 16 ГБайт ОЗУ.
Написано все на Java, исходники открыты, есть хорошо документированный API, потому что часто биоинформатикам удобно встраивать что-то кусками в свой код, так что мы пытаемся сделать ПО максимально удобным в использовании.
Теперь мы подходим к тому, как можно сделать из этих данных диагностику. Уже сейчас все эти эксперименты с секвенированием геномов помогают придумывать какие-то новые лекарства. Но чтобы использовать эти данные как таковые, нужно научиться извлекать из них паттерны.
Это действительно возможно, много статей показывают, что на одни и те же типы инфекций вырабатываются одинаковые CDR3. Это говорит о том, что диагностику построить возможно. Понятно что бывают сложные объекты, которые рождают не очень ярко выраженные паттерны. И чтобы такие паттерны обнаружить, нужна большая статистика, большой набор как контрольных данных (здоровых доноров), так и больных пациентов с известными заболеваниями. Скорее всего, одна клиника не сможет накопить такое количество семплов, чтобы проследить необходимые паттерны для большого спектра заболеваний. И единое место для коллаборации будет способствовать развитию диагностики.
Такая база должна хранить эти данные в какой-то удобной структуре, которая позволяла им делать специфические забросы, необходимые для вычисления паттернов, анализа образцов. Она должна разграничивать доступ, т.к. многие клинические данные нельзя выкладывать в публичный доступ. И также нужно, чтобы эта база данных делала свою работу: вылавливала паттерны данных, а потом искала их в новых данных, постепенно уточняла их через эти же новые данные, улучшала свою производительность.
Такая база настроена на достаточно дальнюю перспективу. С самого начала может быть непонятно, зачем она нужна. Но эти данные сложны сами по себе. Представьте, что вы работаете с сотнями массивов, в каждом из которых по миллиону последовательностей. При этом вам нужно искать между ними закономерности (а они бывают весьма сложными), и ищете вы необязательно полностью эквивалентные CDR3, а, например, похожие. И поиск осуществляется сразу во многих массивах. В общем, находить закономерности — это очень сложно. Нужен какой-то высокоуровневый язык, который позволял бы все это описывать. Сейчас исследователи, которые с этими данными работают, вынуждены каждый раз писать какие-то свои скрипты. На самом деле прослеживать какие-то четкие закономерности хитрее, чем просто нахождение одинаковых клонов одинаковых массивах в одинаковых массивах, сейчас просто не выходит, нет необходимых инструментов. Поэтому, если сделать такую базу, она будет интересна людям, т.к. решит их конкретные исследовательские задачи. И все это должно быть удобно и наглядно, так как часто с этими данными работают биологи, которым графический интерфейс просто необходим. Нужна площадка для коллаборации, так как для действительно интересных результатов нужно много данных, которые стекались бы в одно место из разных источников.
Основная проблема в организации такой общей базы данных — в доставке этих данных от исследователей. Речь идет о сотнях Гбайт, а то и терабайтах. Постепенно это проблема начинает решаться благодаря облачным сервисам. Многие исследователи загружают результаты своей работы на Яндекс.Диск, Dropbox и т.п. Также есть специализированные облачные хранилища. Например, BaseSpace — заточенный специально под секвенаторы Illumina. Т.е. покупая секвенатор, вы автоматически получаете аккаунт в BaseSpace, и данные загружаются в него прямо в процессе секвенирования. Кроме того, у BaseSpace есть удобный API, через который внешние приложения могут получать к нему доступ. Так что его вполне можно использовать в качестве части инфраструктуры.
У нас уже есть некоторый прототип базы данных. Идея такова, что если сырые данные потенциально требуют повторной обработки через какое-то время, то их можно записать на ленту и хранить где-нибудь отдельно. Это самый дешевый способ хранения больших объемов данных. Такие услуги предоставляет, например Amazon Glacier. Обработанные данные можно хранить в реляционной базе данных, но это совершенно необязательно. По сути это неструктурированные данные — списки CDR3. Поэтому их можно хранить как файлы в блочных хранилищах и подгружать их при перестройке структуры базы данных.
Биоинформатикам при вычислении паттернов на низком уровне, скорее всего, понадобятся следующие запросы:
Найти все массивы, которых встречается определенных клон. Найти все клоны, встречающиеся в группе массивов. Различные нечеткие поиски и группировки Чтобы делать это быстро и просто, не писать каких-то собственных алгоритмов, можно использовать PostgreSQL Скорее всего, это не единственный способ, но в прототипе мы воспользовались именно им. В PostgreSQL есть такая вещь как обратный ключ: в полях можно хранить просто массивы интов, соответственно, можно делать запросы типа «дай мне все записи, которые в этих полях содержат цифры 3, 5 и 8». И эти запросы очень быстрые. На картинке ниже изображена упрощенная модель нашего прототипа. Там должна быть еще куча мета-информации, другой побочной информации, которая идет вместе с CDR3. По сути хорошей производительности мы добились за счет того, что в качестве ключей выступали CDR3, а в качестве данных хранился список дата-сетов, в котором этот CDR3 участвовал, в котором он встречался. Поэтому запросы к этому ключу полностью соответствуют тем запросам, которые обычно такая база данных и будет выполнять.Можно поговорить о производительности прототипа более предметно. Для примера возьмем машину с четырехъядерным процессором Intel Core i7 2600 @ 3.8 GHz, 16 Гбайт ОЗУ и софтовым RAID1 из двух SSD по 128 ГБайт каждый. На вход было отправлено 112 массивов, содержащих в общей сложности 45 миллионов колонов. Все это уместилось в 70 Гбайт на SSD. В результате на запросах типа «все клоны из двух массивов» нам удалось добиться производительности в 50 операций в секунду с задержкой около 150 миллисекунд.
В заключение хочется повторить, что аккумуляция данных в одном месте необходима для того, чтобы подобная вещь заработала как диагностика. Сейчас все анализы направлены на то, чтобы определить, есть ли у человека то или иное заболевание. Новый же тип диагностики призван при помощи одного анализа сразу дать ответ, чем именно болен человек. Для этого нужно собрать и обработать очень много данных, и наш прототип показывает, что это вполне реально и не очень ресурсоемко.
